Hypofosfatasi – prematur felling av primære tenner
Hypofosfatasi er en arvelig metabolsk lidelse som kan manifestere seg som tidlig tap av primære tenner. Tilstanden er karakterisert ved en nedsatt aktivitet av enzymet alkalisk fosfatase. Dette påvirker mineralisert vev. Det kliniske bildet varierer sterkt, fra prematurt tanntap til tilnærmet manglende skjelettmineralisering og perinatal død. I tillegg til redusert aktivitet av alkalisk fosfatase har alle pasientene økt utskillelse av aminosyren fosfoetanolamin i urinen og unormal mineralisering av ben og rotsement.
Her presenteres et kasus der en knapt to år gammel jente ble henvist til Klinikk for allmenn odontologi – barn på grunn av prematurt tap av melketenner. Årsaker, mulige differensialdiagnoser samt behandling blir diskutert. Kasuistikken illustrerer også tannlegens betydning ved tidlig diagnose av generelle lidelser.
Hypofosfatasi er en sjelden metabolsk forstyrrelse. De alvorlige former er rapportert å opptre hos omtrent 1/100 000 levende fødte. Sykdommen følger en autosomal arvegang, dominant eller recessiv, der den autosomalt recessive er mest vanlig (1). Tilstanden er karakterisert ved et lavere nivå/aktivitet av enzymet alkalisk fosfatase (ALP) i lever, nyrer og ben. Når alkalisk fosfatase-aktiviteten er nedsatt, akkumuleres pyrofosfat som hemmer mineralisering av ben. Økt utskillelse av fosfoetanolamin og pyrofosfat i urinen er følsomme markører for nedsatt alkalisk fosfatase aktivitet (1,2).
Hypofosfatasi kan skyldes flere ulike punktmutasjoner i genet for vevs-nonspesifikk (lever/ben/nyrer) alkalisk fosfatase på kromosom 1. Selv om alkalisk fosfatase er til stede i de fleste vev, er det spesielt aktivt under mineralisering. Det er lokalisert i cellemembranen der redusert enzymaktivitet bl.a. fører til at pyrofosfat ikke blir hydrolysert til fritt fosfat med konsekvenser for mineraliseringen av ben og tenner (1 – 4).
Tilstanden kan ha mange kliniske uttrykksformer, og alvorlighetsgraden avhenger av gendefektens ekspressivitet/ penetrans. Dette skjer ved at de ulike punktmutasjonene gir ulike aminosyresubstitusjoner, dvs. at gale og forskjellige aminosyrer kommer inn i enzymet. Det er foreløpig beskrevet mer enn 50 ulike mutasjoner. Det kliniske bildet kan således variere fra nærmest total mangel på skeletal mineralisering til kun prematurt tanntap. Jo tidligere symptomene melder seg, desto mer alvorlig er vanligvis tilstanden. Ut ifra alder ved symptomdebut kan sykdommen deles inn i fire ulike former; perinatal, infantil, juvenil og adult.
Perinatal hypofosfatasi opptrer in utero, og barnet er dødfødt eller dør like etter fødselen. Disse barna har dramatiske skeletale forandringer. Infantil hypofosfatasi viser også skeletale avvik, men mindre alvorlige. Symptomene blir som regel diagnostisert innen første halvår etter fødsel. Tilstanden er dødelig i omtrent halvparten av tilfellene.
Juvenil hypofosfatasi blir ofte først diagnostisert av tannlege pga. prematurt tanntap. Symptomene starter gjerne mellom 6 og 24 måneders alder. I tillegg til tidlig tap av primære incisiver kan barnet være lite av vekst, ha rakittforandringer og muskelsvakhet. Når tilstanden først blir diagnostisert i voksen alder, er det vanligvis i forbindelse med prematurt tap av permanente incisiver i over- og underkjeven, og med en anamnese som også avdekker tidlig tap av primære tenner (2,4). Begrepet odontohypofosfatasi brukes i enkelte artikler om tilfeller der man klinisk kun finner tannaffeksjon uavhengig av alder (1,4).
Vanligvis er det individer med den recessivt arvede typen som er hardest rammet, der de to defekte allelene (gener som styrer samme egenskap) kan ha punktmutasjoner som gir ulike aminosyresubstitusjoner i proteinet. De som har dominant hypofosfatasi (bare én vevs-nonspesifikk alkalisk fosfatase allele er defekt), har vanligvis moderate symptomer som tidlig tap av primære tenner (1,3,4).
Pasienten
En 22 mnd. gammel jente ble henvist til Klinikk for allmenn odontologi – barn pga. tidlig tap av primære tenner. Anamnesen ga ingen opplysninger om kjente sykdommer. Pasienten fikk de første temporære incisiver, 71 og 81, ved 2 – 3 mnd. alder. Disse ble felt allerede ved 1-års alder, og 72 og 82 ble felt før pasienten var 1 1/2 år. Alle tenner hadde full rotlengde ved felling. Ved første konsultasjon ved fakultetet hadde pasienten mistet fem primære incisiver; 61, 71, 81, 72 og 82. 51 var mobil (Fig. 1).
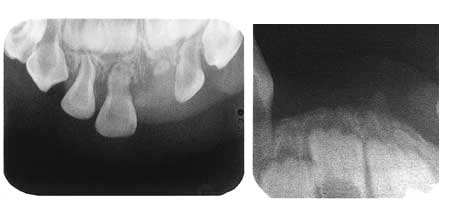
Fig. 1. Jente, 22 mnd. gammel. Fem primære incisiver er felt. Røntgen viser utvidet periodontalspalte på gjenværende incisiver i overkjeven og stort horisontalt tap av alveolært ben i over- og underkjeven. Videre sees utvidete pulpahulrom, men ingen tegn på rotresorpsjon. Apikal rotrest 61, muligens som følge av tidligere traume.
Det forelå ingen gingival inflammasjon. Andre primære molarer var ikke frembrudt. Verken mor eller far hadde, så langt de visste, hatt tilsvarende prematurt tanntap. Pasienten hadde ingen søsken.
Mulige årsaker til det tidlige tanntapet ble vurdert. En form for prepubertal periodontitt kunne ikke umiddelbart utelukkes. Men sett i relasjon til pasientens omfattende tanntap i svært ung alder var en systemisk lidelse mer sannsynlig. Det kliniske og røntgenologiske bildet var dessuten forenlig med den sjeldne metabolske tilstanden hypofosfatasi. Pasienten ble derfor henvist for videre utredning med tanke på slik systemsykdom.
Den medisinske utredningen viste lav serum alkalisk fosfatase (ALP) og nesten åtte ganger høyere nivå av aminosyren fosfoetanolamin i urinen enn øvre referanseområde for barn. Vitamin D-status og kalsiumutskillelse i urin var normal. Av øvrige undersøkelser ble det tatt røntgen av begge håndledd og av thorax. Disse var begge normale.
Screening av genet for vevs-nonspesifikk alkalisk fosfatase i familien viste at begge foreldrene var bærere av tilstanden, og at jenta hadde arvet en mutasjon fra far og en fra mor. Foreldrene hadde også lett forhøyet fosfoetanolamin i urinen, noe som indikerte bærertilstand.
Ny odontologisk undersøkelse ved 2 1/2-års alder viste 9 tapte primære tenner (Fig. 2 og 3). Munnhygienen var god, og det var ingen gingival inflammasjon. Profylaktisk ble bruk av myk børste og riktig børsteteknikk uten tanntråd anbefalt, samt fluortilskudd to ganger daglig.
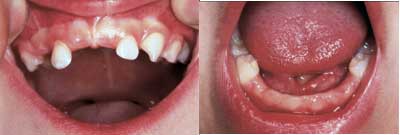
Fig. 2. Klinisk bilde, 32 mnd. gammel. Med unntak av 52 og 62 er overkjevens tenner faste. 53 var eksartikulert etter lett traume, mens 73 og 83 var felt uten klar årsakssammenheng. 52 og 62 har hvite hypomineraliserte flekker incisalt. 74 og 84 er svakt mobile.
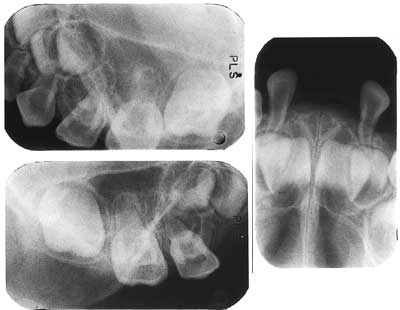
Fig. 3. Røntgen ved 32 måneders alder viser marginalt bentap i overkjeven. Det er anlegg til permanente incisiver, hjørnetenner og 6-årsmolarer, follikeldannelse til første premolar, men foreløpig ingen anlegg til andre premolar.
Diskusjon
Prematur felling av de primære tenner uten at det foreligger en systemsykdom er sjelden.
I dette kasus er det så langt kun det premature tanntapet som har gitt seg klinisk utslag. De intraorale tannbildene viser også redusert benhøyde. Røntgen av begge håndledd og røntgen thorax ble beskrevet som normale, men pasienten blir fulgt opp ved barneavdeling på sykehus med tanke på utvikling av skjelettaffeksjon.
I en større retrospektiv undersøkelse fra Sverige som inkluderte 17 barn, ble det vist at symptomer fra bena var det mest vanlige trekk (hos 16 av de 17 barna) (5). Syv av 15 pasienter viste prematurt tanntap. I det materialet døde fire barn, to før prematurt tanntap kunne registreres.
Den medisinske utredningen i vårt kasus viste at pasienten har en mutasjon i begge sine TNSALP- (tissue non-specific alkaline phosphatase) gener. Hun ser likevel ut til å ha en mild form for sykdommen der det foreløpig bare er påvist tannaffeksjon (odontohypofosfatasi).
Flere årsaksforhold kan spille inn når det gjelder det premature tanntapet. Histologiske undersøkelser som er utført på tapte tenner, har vist både unormal emalje, dentin og sement. Felles synes å være at sementen er affisert i større eller mindre grad hos alle pasienter (5 – 9). Den kan være tynn, eller delvis eller helt fraværende. Det underliggende dentin kan videre vise mineraliseringsforstyrrelser og ha en irregulær overflate med resorpsjonslakuner der den ikke er beskyttet av sement (5,9). Ettersom stabilitet og retensjon av tenner er avhengig av periodontalfibre som er festet både til sement og til alveolært ben, kan irregulær sement eller fravær av sement forklare det dårlige festet og det tidlige tanntapet. Men også cellemetabolismen i periodontalmembranen kan være en medvirkende årsak. Disse cellene har et høyt aktivitetsnivå bl.a. av enzymet alkalisk fosfatase. Det antas at ALP i disse cellene blir regulert på samme måte som i ben og spiller en viktig rolle i hydrolysen av fosforforbindelser til fritt uorganisk fosfat, og at defekt ALP derved påvirker tennenes festemekanisme (10).
En annen teori er at ødeleggelsen av periodontiet skyldes bakterier og er en inflammatorisk prosess på lik linje med periodontitt ettersom mikrober forbundet med periodontitt er påvist subgingivalt hos hypofosfatasipasienter (9,11). Det er imidlertid mer sannsynlig at disse mikrobene er kommet sekundært pga. dårlig periodontalfiberfeste som gjør rotoverflaten mer utsatt for bakteriell invasjon, og at subgingivalt plakk er mer et resultat av enn en årsak til periodontal nedbrytning (8,12). Videre er det ikke alle rotoverflater som viser plakkdannelse ved histologisk undersøkelse. Bakterier synes derfor ikke å være den primære årsak til de forandringer/resorpsjoner som sees i sement/dentin (12).
Hos denne pasienten kunne det observeres utvidete rothinner, men ingen synlig rotresorpsjon. Det er ikke blitt foretatt histologiske undersøkelser av pasientens tenner, men muligheten for at plakk har penetrert subgingivalt er ikke usannsynlig. Det er derfor svært viktig at pasienten har god munnhygiene. At det ikke ble anbefalt bruk av tanntråd, skyldes en potensiell mulighet for å skade de allerede sårbare periodontalfibrene. God munnhygiene er også viktig for å unngå karies på de gjenværende primære molarer. Da det er store pulpahulrom, er det økt fare for pulpaaffeksjon.
Et omfattende prematurt tanntap kan videre forårsake vandringer og plassmangel i det permanente tannsettet. En partiell protese kan i disse tilfellene både hindre mesialvandring samt bedre funksjon og estetikk.
Når det gjelder det permanente tannsettet, blir dette ikke alltid affisert (13). Men når det rammes, er forandringene de samme som i det primære tannsettet (6,9,12). Stedvis mangel på sement gjør tennene utsatt for resorpsjon. Dette kan bl.a. gi seg utslag i trykkresorpsjon fra nabotann i forbindelse med frembrudd (12).
I tillegg til de nevnte patologiske avvik i sement og dentin kan emaljen være hypomineralisert i større eller mindre grad. Dette kan skape problemer ved fyllingsterapi (6). Hos denne pasienten var det kun hypomineraliserte flekker incisalt på overkjevens primære lateraler, men det er på det nåværende tidspunkt ikke mulig å si noe om det permanente tannsettet. Selv om litteraturen først og fremst fremhever forandringene i det primære tannsettet, er det av flere grunner viktig å følge disse pasientene nøye.
Det kliniske bildet med prematurt tanntap åpnet for mulige differensialdiagnoser. Ved lokalisert prepubertal periodontitt affisereres et ulikt antall primære tenner, og gingival inflammasjon er heller ikke der et fremtredende trekk (14). Videre ser man prematurt tanntap ved de arvelige lidelsene Papillon-Lefèvres og Coffin-Lowry syndrom. Ved førstnevnte er imidlertid hyperkeratose på håndflaten og fotsålen karakteristiske trekk (15), og ved sistnevnte er pasienten mentalt retardert og har karakteristiske hovne og koniske fingre (16). Ved neutropeni (også ved cyclisk neutropeni) er det i tillegg til alveolært bentap også gingivitt og sårdannelse. Likeledes vil man se blødning og inflammert gingiva ved Histiocytosis X og leukemi, i motsetning til det man ser hos hypofosfatasipasienter (14). Det vil imidlertid være den medisinske utredningen som fastslår endelig diagnose.
For pasienter med en genetisk lidelse er det av stor betydning å få stilt en diagnose tidlig slik at foreldrene kan få nødvendig genetisk veiledning med tanke på fremtidige barn. I dette tilfellet er mors mutasjon tidligere beskrevet ved moderate infantile og juvenile former for hypofosfatasi hos blandet heterozygote individer. Den er imidlertid også funnet ved en letal form hos et blandet heterozygot individ. Fars mutasjon er ikke beskrevet tidligere. Selv om sykdommen har vist stor fenotypisk variasjon, er det ikke rapportert om interfamiliær variasjon ved samme genotype.
Da prematurt tanntap kan være det første kliniske tegn på sykdommen, spiller tannlegen en nøkkelrolle. En tidlig henvisning for diagnostisk utredning er av vesentlig betydning. Det er videre viktig med en tett oppfølging av pasienten og et nært samarbeid med øvrig helsepersonell.
Konklusjon
Denne pasienten har behov for og får tverrfaglig oppfølging. Hun ser ut til å ha en mild form av sykdommen, selv om hun har mutasjoner i begge sine TNSALP-gener. Hvorvidt skjelettmineraliseringen er nedsatt er forløpig ikke fullt utredet, og hun blir fulgt videre med tanke på skjelettaffeksjon. Odontologisk er det foreløpig kun nødvendig med profylaktiske tiltak ettersom pasienten er kariesfri og snakker bra til tross for tanntapene. Behovet for protetisk erstatning vil bli vurdert fortløpende.
Med en mild variant av lidelsen er det håp om at bare det primære tannsettet affiseres. Vanligvis tapes da kun fronttennene, mens denne pasienten hadde mobile første primære molarer allerede i 2 1/2-års alder. Jo flere primære tenner som går tapt, desto mer alvorlig kan tilstanden synes å være. Det er imidlertid vanskelig ut fra den eksisterende litteratur å si noe om prognosen for det permanente tannsettet på bakgrunn av det orale bildet i så ung alder. Det er den generelle medisinske tilstanden som avgjør alvorlighetsgrad, og til nå har pasienten ingen symptomer på skjelettaffeksjon.
English summary
Hypophosphatasia – premature loss of primary teeth A case report
Hypophosphatasia is a rare, inherited, metabolic disease caused by a defect in the tissue-non-specific alkaline phosphatase (TNSALP) gene. When the activity of this enzyme is reduced, the mineralization process is disturbed. The disease has a varied clinical expression, lethal and non-lethal, and is inherited as either an autosomal recessive or dominant trait. The first and sometimes only clinical sign of the condition is premature loss of primary teeth, referred to as odontohypophosphatasia. Marginal bone loss in combination with hypoplastic or aplastic cementum is the reason for premature tooth loss. This paper presents a girl who had exfoliated five primary incisors at the age of 22 months and nine primary teeth at the age of 32 months. Such abnormal exfoliation requires further investigation and is usually a sign of a systemic disease. Thus, further investigations revealed low level of serum alkaline phosphatase and a raised level of phosphoetaolamine in urine. Screening for mutations in the TNSALP-gene revealed inheritance of one mutation from the mother and one from the father. For the time being there is only a dental manifestation, but the girl has close medical follow-up for possible skeletal manifestations. The parents have also been given genetic counselling. Premature loss of primary teeth is often the reason for parents seeking dental consultation. Early diagnosis is important, and the dentist thus plays an important role in this context.
Referanser
1. Whyte MP. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev 1994; 15: 439 – 61.
2. Hu CC, King DL, Thomas HF, Simmer JP. A clinical and research protocol for characterizing patients with hypophosphatasia. Pediatr Dent 1996; 18: 17 – 23.
3. Hu JC, Plaetke R, Mornet E, Zhang C, Sun X, Thomas HF, et al. Characterization of a family with dominant hypophosphatasia. Eur J Oral Sci 2000; 108: 189 – 94.
4. Chapple IL. Hypophosphatasia: dental aspects and mode of inheritance. J Clin Periodontol 1993; 20: 615 – 22.
5. Lundgren T, Westphal O, Bolme P, Modéer T, Norén JG. Retrospective study of children with hypophosphatasia with reference to dental changes. Scand J Dent Res 1991; 99: 357 – 64.
6. Macfarlane JD, Swart JG. Dental aspects of hypophosphatasia: a case report, family study, and literature review. Oral Surg Oral Med Oral Pathol 1989; 67: 521 – 6.
7. Cheung WS. A mild form of hypophosphatasia as a cause of premature exfoliation of primary teeth: report of two cases. Pediat Dent 1987; 9: 49 – 52.
8. Plagmann HC, Kocher T, Kuhrau N, Caliebe A. Periodontal manifestation of hypophosphatasia. A family case report. J Clin Periodontol 1994; 21: 710 – 6.
9. el-Labban NG, Lee KW, Rule D. Permanent teeth in hypophosphatasia: light and electron microscopic study. J Oral Pathol Med 1991; 20: 352 – 60.
10. Goseki M, Oida S, Takeda K, Ogata Y, Iimura T, Maruoka Y, et al. Identification of bone-type alkaline phosphatase mRNA from human periodontal ligament cells. J Dent Res 1995; 74: 319 – 22.
11. Baab DA, Page RC, Ebersole JL, Williams BL, Scott CR. Laboratory studies of a family manifesting premature exfoliation of deciduous teeth. J Clin Periodontol 1986; 13: 677 – 83.
12. Olsson A, Matsson L, Blomquist HK, Larsson A, Sjödin B. Hypophosphatasia affecting the permanent dentition. J Oral Pathol Med 1996; 25: 343 – 7.
13. Lepe X, Rothwell BR, Banich S, Page RC. Absence of adult dental anomalies in familial hypophosphatasia. J Periodontal Res 1997; 32: 375 – 80.
14. Watanabe K. Prepubertal periodontitis: a review of diagnostic criteria, pathogenesis, and differential diagnosis. J Periodontal Res 1990; 25: 31 – 48.
15. Hall RK. Abnormalities of eruption and resorption, and periodontal disorders in children. In: Pediatric Orofacial Medicine and Pathology. London: Chapman & Hall Medical; 1994: p.127 – 48.
16. Day P, Cole B, Welbury R. Coffin-Lowry syndrome and premature tooth loss: a case report. ASDC J Dent Child 2000; 67: 148 – 50.
Arv; Biokjemi; Kasuistikk; Medfødt sykdom; Tannløshet
Adresse: Anne Skaare, Det odontologiske fakultet, Klinikk for allmenn odontologi – barn, postboks 1109 Blindern, 0317 Oslo. E-post: askaare@odont.uio.no
Artikkelen er fagfellevurdert.
Artikkelen siteres som:
Skaare A, Skogedal N, Myhre AG. Hypofosfatasi – prematur felling av primære tenner. Nor Tannlegeforen Tid. 2002;112:216–9. doi:10.56373/2002-4-3