Den parodontala infektionens lokala konsekvenser
Författare
professor. Institutionen för odontologi, Helsingfors Universitet, Helsingfors, Finland
professor. Karolinska Institutet, Odontologiska Institutionen, Avdelningen för parodontologi, Huddinge, Sverige
Trots att parodontit är en infektionssjukdom har det under de senaste decennierna blivit allt tydligare att patologin finns i kroppens svar på närvaron av bakterier i tandköttsfickan, det så kallade inflammatoriska svaret.
Parodontiten kan förklaras som en allt för kraftig reaktion på de bakterier som finns i tandköttsfickan. I likhet med andra kroniska vävnadsnedbrytande inflammationer, till exempel reumatism eller Crohns sjukdom, är det ännu oklart vad som orsakar överaktiviteten. Teoretiskt finns det i dag flera möjligheter att bekämpa tandköttsfickor och parodontala sjukdomar. En mängd behandlingsformer som säkert kommer att kunna användas i framtiden är också under utveckling. Fortfarande är dock de huvudsakliga vapnen mot skadliga bakterier i munnen de gamla och beprövade: god munhygien, tidig diagnos samt ett effektivt avlägsnande av bakterieplack från tandytorna.
I frisk vävnad råder en noggrant reglerad balans mellan cellerna och vävnadskomponenterna utanför dem. Cellerna kan producera och bryta ner extracellulära strukturella molekyler i rätt förhållande. Hormoner och tillväxtfaktorer har den viktigaste rollen vid regleringen av denna balans. I de följande styckena behandlar vi störningar i den balansen och de förändringar som en parodontal infektion åstadkommer i parodontiets epitel, i bindväven och i alveolarbenet.
Förändringar i munnens mikrobflora leder till lokala förändringar i parodontium
Det är allmänt känt att flertalet parodontala sjukdomar beror på att bakterier växer kring tandköttssulcus. Så småningom förorsakar de kliniska förändringar vi ser i form av rodnande tandkött och ökad blödningsbenägenhet, och som vid parodontit ger upphov till tandköttsfickor. Man vet betydligt mindre om vilken bakterieflora som krävs för att parodontalsjukdomar ska uppstå och utvecklas. I människans tandköttsfickor växer cirka 500 olika bakteriearter. De är organiserade i olika lager, och erbjuder varandra retentionsytor och näring. Det centrala målet med parodontalbehandling är att bryta ned den organisationen och avlägsna retentionsytor som tandsten och ofysiologiska anatomiska strukturer på tänderna.
Frågan om vilka bakteriearter som hänger samman med parodontala sjukdomar har ägnats mycket forskning. Även om det har skett många framsteg på det området är vår kunskap om vilka arter som är nödvändiga för parodontalsjukdomarnas patogenes och vilka som annars bara finns med fortfarande bristfällig. Man vet att anaeroba gramnegativa och rörliga bakteriearter, till exempel spiroketer, dominerar i den subgingivala placken. Man har också kommit fram till att vissa bakteriearter kan klassificeras som patogena enligt följande kriterier:
De förekommer i större mängd vid parodontala sjukdomar än i friskt parodontium.
Om man eliminerar dem förbättras den parodontala hälsan.
Hos försöksdjur ger de upphov till en vävnadsreaktion som främjar nedbrytning av parodontiet.
De har virulensfaktorer som in vitro har visat sig ha skadliga effekter på celler och vävnader.
Sådana arter är bland annat Porphyromonas gingivalis, Actinobacillus actinomycetemcomitans och Treponema denticola (1). Man vet att de alla producerar många faktorer, till exempel lipopolysackarid, peptidoglykan, kväveföreningar och enzymer, som kan vara delaktiga då parodontala sjukdomar utvecklas. Det är knappast ett sammanträffande att P. gingivalis och T. denticola har en kraftig proteolytisk arsenal med vilken de kan förstöra vissa faktorer i vävnadernas försvar, till exempel immunoglobuliner, komplementkomponenter och fibrin. På det sättet kan de undvika försvarsreaktioner och växa i tandköttsfickorna. Också många andra bakteriearter i munnen har virulenta varianter som kan medverka till uppkomsten av parodontit.
Förändringar i tandköttets epitel ger upphov till tandköttsfickor
Tandköttets epitel är det första målet för bakteriekolonierna i sulcus (2). Epitelet som bildar fickans laterala vägg är parakeratiniserat och motstår bakteriernas inverkan ganska väl. Kontaktepitelet är ett okeratiniserat och relativt permeabelt flerskiktat epitel. En situation där ett icke keratiniserat epitel är fäst vid en inert hårdvävnad finns inte på något annat ställe i organismen. Det ställer naturligtvis specifika krav på försvaret mot bakterierna i munnen.
Kontaktepitelet är långt ifrån försvarslöst. Det har i själva verket ett mångsidigt system för att kämpa mot bakterier. Dess förnyelsehastighet är sällsynt hög. Hos apor är förnyelsehastigheten cirka fem dagar, ungefär dubbelt så snabb som hos det orala epitelet. Eftersom det snabbt förnyas och lossnar från sulcusregionen för det snabbt bort bakterier som är fästa vid epitelceller. Undersökningar har på senare tid visat att kontaktepitelet har flera specifika försvarsmetoder. Det innehåller lysosomer som kan förstöra bakterier som trängt in i cellerna. Både sulcus- och kontaktepitelet producerar defensiner, antimikroba peptider som mycket effektivt förstör många olika bakterier (3). Leukocyter, i första hand neutrofila, migrerar genom kontaktepitelet och utgör en viktig länk i parodontiets försvar. Cellerna i kontaktepitelet producerar kemotaktiska ämnen, till exempel interleukin-8, som effektivt lockar till sig leukocyter från de talrika blodkärlen under epitelet. Även om tandköttet är kliniskt friskt innehåller kontaktepitelet en anmärkningsvärt stor mängd neutrofiler. Kontaktepitelets försvarslinje utgörs av de inflammationsfaktorer som strömmar med gingivalvätskan. Vissa av dem härstammar från serum och produceras av neutrofiler, makrofager, lymfocyter och plasmaceller. Förutom av plasmaceller som cirkulerar i blodet produceras antikroppar mot bakterier lokalt i tandköttsvävnaden. Kontaktepitelet har eventuellt en roll i denna lokala antikroppsproduktion. Tandköttets försvar kan anses fungera överraskande väl med tanke på att det hela tiden finns bakterier i sulcus som är beredda att växa in i tandköttsvävnaderna. Vissa av arterna har förmåga att röra sig och tränga in i vävnaden. Det är ändå mycket sällan bakterier i någon betydande utsträckning lyckas tränga igenom tandköttsepitelet till bindväv och blodomlopp.
För att vi ska förstå hur parodontala sjukdomar uppstår måste vi först veta hur tandköttet är fäst vid tanden i ett friskt parodontium: Fästet består av hemidesmosomer på ytan till kontaktepitelet och ett basalmembran på tandytan, kallad tandköttets inre basalmembran (det yttre basalmembranet finns mellan bindväv och epitel). Morfologiskt liknar det inre basalmembranet andra basalmembran men biokemiskt är det unikt. Det saknar de typiska basalmembranstrukturerna (kollagen typ IV och laminin-1). Däremot är laminin-5 en viktig komponent. Här binds epitelcellernas receptorer (integrinerna) i hemidesmosomer (2).
Den patofysiologiska mekanismen som leder till att tandköttsfickor uppstår är ännu inte klarlagd. Många faktorer kan inverka på processen. Först och främst kan proteolytiska bakterier som P. gingivalis och T. denticola direkt bryta sönder laminin-5 och epitelfästet. Dessutom innehåller ett flertal kroppsegna celler, till exempel neutrofiler, fibroblaster och epitelcellerna själva enzymer som också kan bryta ned denna fog. Neutrofiler innehåller flera hydrolytiska enzymer som tillsammans kan bryta ned alla olika vävnadskomponenter i parodontiet. I själva verket härstammar flertalet av de proteolytiska enzymerna i gingivalvätskan från neutrofiler. Bakterierna aktiverar också många andra celler, till exempel epitelceller, fibroblaster och lymfocyter. Dessa celler börjar producera enzymer som bryter ned vävnaderna. Bakteriernas inverkan på uppkomsten av tandköttsfickor utgörs alltså av de direkta effekter de utövar på gingivalvävnaden och den skadliga kedjereaktion som får tandköttet att lossna från tandytan.
När tandköttsfickor bildas sker det många förändringar i kontaktepitelet (tabell 1, figur 1). Epitelcellernas funktion rubbas av många ämnen och inflammationsfaktorer som härstammar från bakterier. Många ämnen som bakterierna producerar är toxiska. De leder till ospecifik försvagning av cellfunktionerna och celldöd. Sådana ämnen är till exempel kväveföreningar som speciellt vissa anaeroba bakterier avger. Dessutom söker sig många ämnen som härstammar från bakterier specifikt till receptormolekyler på cellernas yta och orsakar på det sättet störningar i cellernas signaltrafik. Cellernas funktion är beroende av en mängd signalsystem. När de aktiveras kan cellfunktionerna antingen bli långsammare eller stimuleras (figur 2). Som exempel kan man nämna iakttagelsen att lipopolysackarider och stressproteiner i små mängder på bakteriernas yta ökar cellernas delning och rörelse medan höga halter inhiberar dem. Ämnenas effekt på cellerna är alltså beroende av från vilka bakterier de härstammar, vid vilka receptorer de binds och i hurdana koncentrationer de förekommer i tandköttsfickorna. En av de bäst kända signalrutterna är de reaktioner som går under namnet MAP-kinas (4). De består av flera enzymkedjor i serie efter varandra. Deras uppgift är att öka de effekter en liten signal har inom cellen. En MAP-kinasväg kan öka celldelningen när den aktiveras, andra förorsakar programmerad celldöd eller apoptos. Apoptosens betydelse ligger bland annat i att infekterade celler och de bakterier som är bundna vid den kan transporteras bort av makrofager, snyggt hoppackade, utan att de orsakar onödiga problem i sin omgivning.
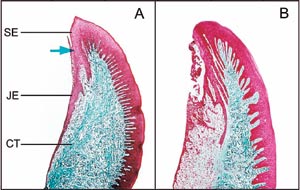
Figur 1. Histologisk jämförelse mellan frisk (A) och inflammerad (B) gingiva. Den friska gingivan ansluter sig tätt till tanden med kontaktepitel eller s.k. junktionalt epitel (JE) ända till botten av sulcus (pilen). Bindväven är fylld av kollagenfibrer som färgats blå (CT). Den inflammerade gingivan är inte fast förankrad vid tandytan. Epitelet har börjat växa in i bindväven, och kollagenfibrerna har brutits ned under epitelet.
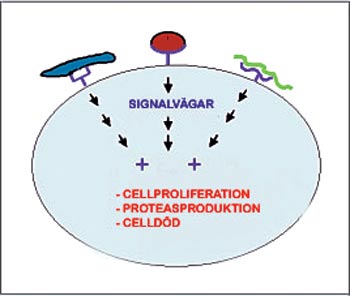
Figur 2. När bakterier fäster sig vid receptorerna på epitelcellernas yta aktiveras enzymatiska signalreaktioner som leder till förändringar i vissa cellfunktioner, till exempel celldelningen och den syntetiska aktiviteten. Det kan också leda till celldöd (apoptos).
Epitelcellerna aktiveras |
Epitelets permeabilitet ökar |
Epitelcellerna avger försvarsfaktorer, exempelvis ämnen (IL-8) som lockar till sig leukocyter |
Antalet neutrofiler (PMN) i epitelet ökar |
Strömningen av gingivalvätska genom epitelet ökar |
Epitelcellernas delningshastighet och lösgörelse från sulcus ökar |
Kontaktepitelet lossnar från tandytan och förändras till fickepitel |
Cellerna i fickepitelet avger enzymer som gör det möjligt för dem att växa in i underliggande bindväv |
En av förändringarna i cellerna som förorsakas av att signalvägarna aktiveras är epitelcellernas enzymavsöndring. Den leder till att bindningarna mellan cellerna och de pericellulära vävnadsmaterialen faller sönder och tandköttsfickans epitels permeabilitet ökar. Då kan leukocyterna lättare röra sig mot bakterierna, men samtidigt underlättas diffusionen av ämnen från bakterierna till tandköttet. Också blodkärlen under tandköttets epitel får större permeabilitet och strömningen av gingivalvätska från vävnaderna till tandköttsfickan mångfaldigas jämfört med frisk vävnad. Det är betydelsefullt eftersom ämnen från serumet och övrig materia främst strömmar i riktning från vävnaderna. Det gör det i sin tur svårare för ämnen från bakterierna att tränga in i parodontiet.
Epitelceller kan med hjälp av de enzymer de avsöndrar tränga in i den underliggande bindväven. Man har kunnat påvisa att tandköttsfickans epitelceller som aktiverats av parodontit avsöndrar ett speciellt potent kollagenas som bryter ned fibrerna i parodontiet (figur 3). På det sättet deltar alltså fickornas epitel aktivt i nedbrytningen av parodontiet och bildandet av tandköttsfickor (5).
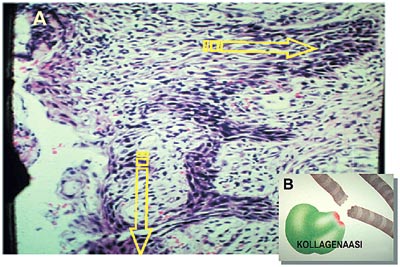
Figur 3. Det aktiverade fickepitelet (A) prolifererari lateral och apikal riktning. Det avger enzymer som förstör bindväven i omgivningen, till exempel kollagenas (B).
Förändringar i tandköttets bindväv
Gingivit är en av de vanligaste kroniska inflammationerna hos människan. Tre till fyra dygn efter det att bakterier samlats i tandköttsfickan och startat en inflammationsreaktion börjar bindväven att brytas ned. Nedbrytningen inleds i de kollagena fiberbuntar som omger blodkärl. Cirka 70 procent av kollagenet i den inflammerade bindväven går förlorad. Nedbrytningen orsakas huvudsakligen av proteaser som frisätts från kroppsegna celler till exempel PMN-celler och fibroblaster. Denna första kollagenförlust leder till att gingivan förlorar sin strama struktur och blir svullen, mjuk och röd. Vanligtvis uppstår sedan en balans mellan nedbrytningen och nybildningen och inflammationen begränsas till gingivan. Om kollagennedbrytningen fortsätter djupare in i vävnaden och når de scharpeyska trådarna, det vill säga de översta kollagena fiberbuntarna i parodontalligamentet övergår gingiviten i en parodontit.
Förändringar i parodontalligament
Karaktäristiskt för parodontiten är nedbrytningen av parodontalligamentet, det vill säga de kollagena fibrer som fäster tanden till alveolarbenet. Det pågår ständigt en omsättning av kollagenet i parodontalligamentet. En hög kollagenomsättning är troligen en förutsättning för att kunna anpassa sig till de krafter som parodontiet utsätts för genom till exempel tuggning. Parodontalligamentets trådar består till största delen av kollagen typ I, samma kollagen som finns i ben och senor. Detta kollagen bryts huvudsakligen ned av två specifika enzymer så kallade kollagenaser, matrixmetalloproteas-1 (MMP-1) och MMP-8 (6). MMP-1 produceras och frisätts av fibroblaster och deltar i den naturliga omsättningen av kollagen. MMP-8 frisätts från PMN-celler i samband med cellens vandring från blodbanan genom vävnaden. Mängden frisatt MMP-8 återspeglar mängden PMN-celler och därmed inflammationsgraden i området. MMP-1 är det dominerande kollagenaset inne i tandköttet medan MMP-8 dominerar i det inflammatoriska eksudatet i tandköttsfickan (7 – 9). Båda dessa kollagenaser frisätts i en latent, det vill säga icke aktiv form, för att sedan aktiveras utanför cellen. Aktiveringen sker via andra proteaser eller via reaktiva syreradikaler. Kroppen har naturliga skyddsmekanismer mot kollagenaser. Tissue inhibitors of matrixmetalloproteinases (TIMP) är ett antal specifika hämmare av kollagenaser. Även den non-specifika proteashämmaren a-2-makroglobulin hämmar kollagenaser. Betydelsen av eventuella förändringar i koncentration och aktivitet av TIMP för utvecklingen av parodontit är inte så väl klarlagt (10). Vissa studier tyder dock på att koncentrationerna av TIMP är lägre vid parodontal sjukdom (11).
Den avgörande skillnaden mellan den normala och funktionella inflammationsreaktionen gingivit och den vävnadsnedbrytande parodontiten tycks inte vara mängden frisatt kollagenas utan hur mycket av detta som aktiveras och hur mycket som sedan hämmas av TIMP. Det finns ett antal studier som visar att patienter med parodontit har mer aktivt MMP-8 dels i jämförelse med patienter med enbart gingivit dels jämfört med friska (12, 13). Höga koncentrationer av latent kollagenas har däremot förknippats med gingivit (12). Sammanfattningsvis kan sägas att hög kollagenasaktivitet och lite TIMP är förknippat med aktiv parodontit och det motsatta förhållandet med parodontal hälsa.
Inflammationsceller
Parodontit är en infektionssjukdom. Trots detta har det under de senaste decennierna blivit allt tydligare att själva patologin står att finna i kroppens svar på bakterienärvaron i tandköttsfickan, det inflammatoriska svaret. Även i kliniskt helt inflammationsfria fickor pågår en ständig vandring av vita blodkroppar från blodkärl, genom bindväv och fickepitel ut i tandköttsfickan. Mer bakteriebeläggningar leder till en kraftigare inflammationsreaktion. Men även på ytor helt utan plackkontroll infinner sig en balans mellan bakterier och kroppens försvar, det blir en kronisk gingivit som inte vidare bryter ned de parodontala vävnaderna. Hos en mindre del av befolkningen infinner sig inte denna balans utan att gingiviten blir vävnadsnedbrytande och övergår därmed i en parodontit. Vad som gör att den gingivala inflammationen blir vävnadsnedbrytande är ännu oklart. Trots intensiv forskning under det senaste decenniet har det inte gått att finna en specifik markör för den vävnadsnedbrytande inflammationen. Alla cytokiner, proteaser, celltyper som finns i parodontitlesionen tycks också finnas i gingiviten. Av någon anledning är den delikata balansen mellan pro- och anti-inflammatoriska cytokiner, proteaser och antiproteaser samt oxidanter och antioxidanter rubbad vid parodontit (14, 15).
Vilka celler som är den egentliga orsaken till den överdrivna inflammationsreaktionen är oklart. Det kan vara alltifrån epitelcellerna i fickan till makrofager och t-lymfocyter i bindväven som överreagerar.
Neutrofila granulocyter eller PMN-celler är näst efter mekaniska barriärer som hud och slemhinnor kroppens främsta försvar mot bakterieinvasion. Vid gingivit och parodontit ansamlas stora mängder PMN-celler i tandköttsfickan och i bindväven alldeles under fickepitelet. Dessa cellers uppgift är att hindra bakterierna att tränga djupare in i vävnaden. Ansamling av PMN-celler är en normal och funktionell reaktion som är en förutsättning för oral hälsa (16). Patienter som saknar PMN-celler, agranulocytos, har ofta en mycket grav tandlossning. PMN-cellernas skydd mot bakterieinvasion fungerar även hos patienter med parodontit. Dessa patienter har inte en spridning av parodontala bakterier djupare in i vävnaden.
En förklaring till att inflammationen hos patienter med parodontit är allt för kraftig och långvarig skulle kunna vara att PMN-cellerna hos dessa patienter är defekta och inte förmår att döda bakterierna tillräckligt effektivt. Mikroorganismerna skulle alltså kunna finnas kvar i vävnaden längre tid och därmed underhålla inflammationen. Det finns ett antal studier som tyder på detta, speciellt vid aggressiv parodontit. Dock visar senare studier att tandlossning inte i första hand orsakas av en underfunktion hos PMN-cellen utan av en allt för stark reaktion hos dessa celler. I sin kamp mot bakterierna frisätter PMN-cellerna såväl proteaser som reaktiva syreradikaler. Detta sker både vid gingivit och parodontit. Det tycks dock som om parodontitpatienternas PMN-celler frisätter dessa ämnen så snabbt och i stor mängd att kroppens naturliga försvar, antiproteaser och antioxidanter, inte förmår att hindra en vävnadsnedbrytning. Perifera (celler som cirkulerar i blodomloppet) PMN-celler från patienter med parodontit som stimuleras in vitro producerar mer reaktiva syreradikaler och släpper ut mer av det proteinspjälkande enzymet elastas än PMN-celler från personer utan parodontit (17, 18). Också studier av gingivalexsudat (GCF) visar på en lokal hyperaktivitet hos PMN-cellerna. GCF-prover från patienter med parodontit innehåller mer elastas och mer b-glukuronidas, som båda är enzymer som frisätts i samband med att PMN-celler fagocyterar bakterier (19 – 21).
Längre in i bindväven samlas monocyter/makrofager som också har ett stort inflytande på inflammationsreaktionens intensitet och duration. På motsvarande sätt som för PMN-cellen finns det studier som visar att perifera monocyter från patienter frisätter mer proinflammatoriska cytokiner (interleukin-1b och TNFa) och prostaglandin E2 (22, 23). Även lokalt i det inflammerade tandköttet sker det en kraftigare frisättning av IL-1b och TNFa hos patienterna med parodontit än hos personer med enbart kronisk gingivit (24).
Cytokiner är lösliga signalsubstanser som produceras och frisätts av celler och som styr andra cellers funktioner och aktivitet. De flesta cytokiner påverkar ett flertal olika celler lokalt. Cytokiner kan ha olika effekter på olika celler och vid olika koncentrationer. I stora drag kan cytokiner delas in i pro-inflammatoriska eller anti-inflammatoriska. Exempel på pro-inflammatoriska cytokiner är interleukin (IL)-1 b, IL-6 och tumour necrosis factor a (TNFa). Anti-inflammatoriska cytokiner som kan vara av betydelse i parodontiet är IL-4 och IL-10. Vid parodontit tycks det vara en dominans av pro-inflammatoriska cytokiner främst IL-1b och TNFa (25).
T-lymfocyter, i första hand T-hjälparceller har genom sin frisättning av cytokiner ett stort inflytande på immunologiska och inflammatoriska reaktioner i den parodontala lesionen. Dessa celler delas in i tre grupper Th1, Th2 och Th3. Grupperna skiljer sig genom att de frisätter olika cytokiner. Det finns inte några publicerade studier om Th3 cellens roll i parodontiten. Däremot finns det ett stort antal studier som försökt kartlägga betydelsen av Th1 och Th2. Resultaten är motstridiga men de flesta studier pekar på att det föreligger en viss Th2 dominans (26). Hur en eventuell obalans mellan Th1 och Th2 påverkar parodontitens utveckling är oklart.
Figur 4 visar hur närvaron av bakterier i tandköttsfickan kan leda till vävnadsnedbrytning och i förlängningen tandförluster (27).
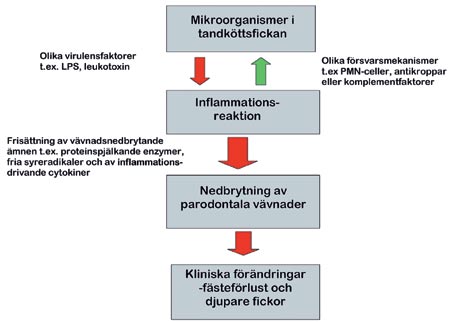
Figur 4. Schematisk beskrivning av de reaktioner som leder från bakteriekolonialisationen i tandköttsfickan till fäste- och tandförlust. Bakteriernai fickan orsakar en försvarsreaktion som effektivt hindrar bakterierna från att tränga in i kroppen men som samtidigt frisätter ämnen som förstör den omgivande vävnaden. Detta leder på sikt till fästeförlust och fördjupade fickor. Dessa förändringar leder i sin tur till förändringar i mikro-floran, det vill säga det blir fler och mer anaeroba bakterier.
Konsekvenser i alveolarbenet
Det pågår en ständig omsättning av det alveolära benet. Vid parodontal hälsa råder det balans mellan nybildning och nedbrytning av ben vilket leder till att benet förnyas samtidigt som benvolymen förblir konstant. Bildandet av osteoblaster som i sin tur bildar ben styrs av en serie av reaktioner som inbegriper så väl hormoner som cytokiner och tillväxtfaktorer (28).
Nedbrytning av ben kräver en specialiserad cell, osteoklasten, som från början är en monocyt/makrofagliknande cell ursprungligen från benmärgen. Bildandet, aktiveringen och även inaktiveringen av osteoklaster beror på ett stort antal ämnen som samverkar respektive motverkar varandra. Pro-inflammatoriska cytokiner, främst interleukin-1b (IL-1b) och tumour necrosis factor a (TNFa) har en avgörande inverkan på osteoklasternas aktivitet (29). Dessa cytokiner stimulerar inte bara bildandet av osteoklaster utan stimulerar också osteoklastaktivitet. Il-1b hämmar också apoptos hos dessa celler. Olika arakidonsyra metaboliter har en inverkan på benomsättningen. Detta gäller särskilt prostaglandiner som kan aktivera osteoklaster, troligen genom att potentiera effekten av olika tillväxtfaktorer.
Alveolarbenet skiljer sig inte på något avgörande sätt från övrigt ben i kroppen men det är beroende av närvaron av tänder och parodontium. Följaktligen resorberas delar av det alveolära benet efter tandextraktioner.
Benförlusten vid parodontit är antagligen en konsekvens av att parodontiet brutits ned och att benets funktion därmed har upphört samt av att den ständiga och överdrivna inflammationen vid parodontit frisätter stora mängder av TNFa och Il-1b som i sin tur aktiverar osteoklaster. I båda fallen måste bennedbrytningen ses som ett sekundärt fenomen som uppstår på grund av en överdriven inflammationsreaktion i de övriga parodontala vävnaderna och inte som ett primärt fenomen även om benförlust på röntgen tillsammans med klinisk fästeförlust är de främsta tecknen på en parodontal sjukdom.
Sammanfattningsvis verkar parodontiten vara en alltför kraftig reaktion på de bakterier som finns i tandköttsfickan. I likhet med andra kroniska vävnadsnedbrytande inflammationer, till exempel reumatism eller Crohns sjukdom, är det ännu oklart vad som orsakar överreaktiviteten.
Hur kan vi då bekämpa tandköttsfickor och parodontalsjukdomar? Teoretiskt har vi flera möjligheter. Vi kan applicera antibakteriella preparat med brett spektrum i tandköttsfickorna. Det finns redan klorhexidinhaltiga chips på marknaden som är avsedda för detta. I framtiden kan man kanske använda defensiner för att öka tandköttsfickornas naturliga försvar. Eventuellt kommer man att kunna förhindra att bakterierna fäster sig vid epitelcellerna och de signaler som följer av det med selektiva läkemedel. Man kan också motverka effekterna av de skadliga enzymer epitelcellerna producerar med enzyminhibitorer. Det utvecklas en mängd nya behandlingsformer och man kommer säkert att ha nytta av dem i framtiden. I dag är de huvudsakliga vapnen mot skadliga bakterier i munnen de gamla och beprövade: viktigast är att sköta munhygienen effektivt, det är också viktigt att diagnostisera parodontalsjukdomar tidigt och avlägsna bakterieplack effektivt från tandytorna så att parodontalsjukdomarna kan behandlas så tidigt som möjligt.
English summary
Uitto V-J, Gustafsson A.
Local consequences of periodontal infections
36 – 41.
Certain pathogenic bacteria in the oral cavity are known to initiate immunological and inflammatory reactions in the periodontium, leading to changes in the epithelium, connective tissue and bone that are characteristic of periodontal diseases. Clinically, these changes involve gingival inflammation, formation of gingival pockets and loss of the normal structure and function of periodontium.
Periodontitis is characterised by destruction of the periodontal tissues, primarily the collagen fibres in the periodontal ligament and secondarily, the alveolar bone. This tissue destruction is mediated by mainly endogenous proteinases, such as matrix metalloproteinase-8 and elastase, in conjunction with reactive oxygen species.
Patients with periodontitis seem to have a hyperinflammatory pattern with excessive inflammatory reaction to the presence of bacteria in the gingival pocket. To protect the body from bacterial invasion, an inflammatory reaction is both functional and necessary. During the fight with the bacteria in the gingival pocket tissue, degrading substances, such as reactive oxygen species and proteases, are released into the surrounding tissues also in patients with gingivitis alone. These substances are normally neutralised by antioxidants and antiproteases in the tissues, but in patients with periodontitis this balance seems to be disturbed. A basically protective reaction becomes tissue destructive.
The first target of periodontopathogenic bacteria is the junctional epithelium. Epithelial cells respond to the microbial attack by either activating or inhibiting cell functions, depending on the type and dose of virulence determinants of the infecting microbes. Junctional epithelium has its own versatile antimicrobial mechanism, which we have recently begun to understand better. This system includes increased cell proliferation, production of antimicrobial substances, such as lysozyme and defensins, and attraction of traditional defence cells, notably neutrophils, to the infected tissue.
Bacteria have developed various ways to manipulate eukaryotic cells in order to avoid elimination by the host response and to create an environment for growth. As a consequence of this epithelial-bacterial interaction, junctional epithelial cells start to proliferate and grow towards the underlying connective tissue, both apically and laterally. Degradation of the pericellular matrix by proteinases secreted by epithelial cells and transmigrating neutrophils partly regulates the rate of epithelial growth. Loss of attachment of the junctional epithelium from the internal basal lamina on the tooth surface leads to formation of gingival pockets.
Concomitant with the changes in epithelium, the subepithelial connective tissue and alveolar bone change drastically during periodontal infection. Continuing infection leads to an excess of proinflammatory cytokines, such as interleukin-1 and tumor necrosis factor-alfa, in periodontal tissues. In the gingiva and periodontal ligament, proteolytic enzymes, including collagenases and elastases, are secreted from leukocytes and activated fibroblasts and lead to destruction of periodontal attachment. Firm connective tissue is replaced by loose tissue infiltrated by leukocytes.
Increased vasculature and epithelial ulcerations increase the tendency of the gingiva to bleed. In alveolar bone the osteoclasts are activated, causing resorption of marginal bone. Not until a certain amount of alveolar bone has been lost can periodontal disease be seen in the radiographical examination. At earlier stages of the disease, however, the inflammatory mediators and proteolytic enzymes can be measured in gingival crevicular fluid. In the future, tests of crevicular fluid will provide accurate and practical means to detect early signs of periodontitis. In addition, efficient methods to prevent subgingival plaque growth and tissue destruction will be available. Meanwhile, periodontal probing and removal of bacterial plaque from tooth surfaces provide the best means of promoting periodontal health.
Referenser
1. Socransky SS, Haffajee AD. Dental biofilms: difficult therapeutic targets. Periodontol 2000 2002; 28: 12 – 55.
2. Pöllänen MT, Salonen JI, Uitto V-J. Structure and function of the tooth-epithelial interface in health and disease. Periodontol 2000 2003; 31: 12 – 31.
3. Dale BA, Krisanaprakornkit S. Defensin antimicrobial peptides in the oral cavity. J Oral Pathol Med 2001; 30: 321 – 7.
4. Zhang L, Pelech SL, Mayrand D, Grenier D, Heino J, Uitto V-J. Bacterial heat shock protein-60 increases epithelial cell proliferation through the ERK1/2 MAP kinases. Exp Cell Res 2001; 266: 11 – 20.
5. Uitto V-J, Airola K, Vaalamo M, Johansson N, Putnins EE, Firth JD, et al. Collagenase-3 (matrix metalloproteinase-13) expression is induced in oral mucosal epithelium during chronic inflammation. Am J Pathol 1998; 152: 1489 – 99.
6. Birkedal-Hansen H. Role of matrix metalloproteinases in human periodontal diseases. J Periodontol 1993; 64: 474 – 84.
7. Uitto V-J, Overall CM, McCulloch C. Proteolytic host cell enzymes in gingival crevicular fluid. Periodontol 2000 2003; 31: 71 – 104.
8. Ingman T, Könönen M, Konttinen YT, Siirilä HS, Suomalainen K, Sorsa T. Collagenase, gelatinase and elastase activities in sulcular fluid of osseointegrated implants and natural teeth. J Clin Periodontol 1994; 21: 301 – 7.
9. Chen HY, Cox SW, Eley BM, Mäntylä P, Rönka H, Sorsa T. Matrix metalloproteinase-8 levels and elastase activities in gingival crevicular fluid from chronic adult periodontitis patients. J Clin Periodontol 2000; 27: 366 – 9.
10. Hayakawa H, Yamashita K, Ohwaki K, Sawa M, Noguchi T, Iwata K et al. Collagenase activity and tissue inhibitor of metalloproteinases-1 (TIMP-1) content in human whole saliva from clinically healthy and periodontally diseased subjects. J Periodont Res 1994; 29: 305 – 8.
11. Haerian A, Adonogianaki E, Mooney J, Manos A, Kinane DF. Effects of treatment on gingival crevicular collagenase, stromelysin and tissue inhibitor of metalloproteinases and their ability to predict response to treatment. J Clin Periodontol 1996; 23: 83 – 91.
12. Lee W, Aitken S, Sodek J, McCulloch CA. Evidence of a direct relationship between neutrophil collagenase activity and periodontal tissue destruction in vivo: role of active enzyme in human periodontitis. J Periodont Res 1995; 30: 23 – 33.
13. Romanelli R, Mancini S, Laschinger C, Overall CM, Sodek J, McCulloch CA. Activation of neutrophil collagenase in periodontitis. Infect Immun 1999; 67: 2319 – 26.
14. Page RC, Schroeder HE. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab Invest 1976; 34: 235 – 49.
15. Degre M. Cytokines and bacterial infections. Biotherapy 1996; 8: 219 – 28.
16. Delima AJ, Van Dyke TE. Origin and function of the cellular components in gingival crevice fluid. Periodontol 2000 2003; 31: 55 – 76.
17. Figueredo CMS, Gustafsson A, Åsman B, Bergström K. Increased release of elastase from in vitro activated peripheral neutrophils in patients with adult periodontitis. J Clin Periodontol 1999; 26: 206 – 11.
18. Buchmann R, Hasilik A, Van Dyke TE, Lange DE. Amplified crevicular leukocyte activity in aggressive periodontal disease. J Dent Res 2002; 81: 716 – 21.
19. Cox SW, Eley BM. Cathepsin B/L-, elastase-, tryptase-, trypsin- and dipeptidyl peptidase IV-like activities in gingival crevicular fluid. A comparison of levels before and after basic periodontal treatment of chronic periodontitis patients. J Clin Periodontol 1992; 19: 333 – 9.
20. Figueredo CMS, Gustafsson A. Activity and inhibition of elastase in GCF. J Clin Periodontol 1998; 25: 531 – 5.
21. Lamster IB, Holmes LG, Gross KB, Oshrain RL, Cohen DW, Rose LF et al. The relationship of beta-glucuronidase activity in crevicular fluid to clinical parameters of periodontal disease. Findings from a multicenter study. J Clin Periodontol 1994; 21: 118 – 27.
22. Shapira L, Soskolne WA, Sela MN, Offenbacher S, Barak V. The secretion of PGE2, IL-1b , IL-6, and TNFa by adherent mononuclear cells from early onset periodontitis patients. J Periodontol 1994; 65: 139 – 46.
23. Offenbacher S, Odle BM, Van Dyke TE. The use of crevicular fluid prostaglandin E2 levels as a predictor of periodontal attachment loss. J Periodont Res 1986; 21: 101 – 12.
24. Figueredo CMS, Ribeiro MSM, Fischer RG, Gustafsson A. Increased interleukin-1bconcentration in gingival crevicular fluid as a characteristic of periodontitis. J Periodontol 1999; 70: 1457 – 63.
25. Howells GL. Cytokine networks in destructive periodontal disease. Oral Dis 1995; 1: 266 – 70.
26. Kinane DF, Lappin DF. Clinical, pathological and immunological aspects of periodontal disease. Acta Odontol Scand 2001; 59: 154 – 60.
27. Page RC, Kornman KS. The pathogenesis of human periodontitis: an introduction. Periodontol 2000 1997; 14: 9 – 11.
28. Sodek J, McKee MD. Molecular and cellular biology of alveolar bone. Periodontol 2000; 24: 99 – 126.
29. Reddy SV, Roodman GD. Control of osteoclast differentiation. Crit Rev Eukaryot Gene Expr 1998; 8: 1 – 17.
Søkeord for nettversjon: www.tannlegetidende.no: Gingivitt; Mikrobiologi; Periodontitt, marginal.
Adress: Veli-Jukka Uitto, professor, Institutionen för odontologi, Helsingfors Universitet, POB 41, FIN-00014 Helsingfors Universitet, Finland. E-post: jukka.uitto@helsinki.fi