Humana papillomvirus och orala infektioner
Författar
professor. Avdelningen för oralpatologi och oralradiologi, Odontologiska institutionen, Medicinska fakulteten, Åbo universitet, Finland
Humana papillomvirus (HPV) är små DNA-virus som infekterar basalcellerna och eventuellt också epitelstamcellerna i epitelvävnader. Antalet identifierade HPV-typer uppgår för närvarande till 106. Baserat på kliniskt utfall kan HPV delas in i benigna och maligna varianter. Och efter sin målvävnad kan man också dela in dem i virustyper som infekterar hud och sådana som infekterar slemhinnor. I den internationella virusklassifikationen räknades papillomvirus som en separat grupp först år 2004.
HPV är den enskilt viktigaste riskfaktorn för cancer i livmoderhalsen, men viruset orsakar också andra former av skivepitelscancer. Man känner ganska väl till hur de processer som HPV-infektionen leder till på cellnivå påverkar den infekterade värdcellen, hur HPV först gör cellen odödlig och sedan omvandlar den till en cancercell. Av de sex tidiga HPV-generna är E6 och E7 särskilt tydligt relaterade till cancer. Dessa gener binder till ett flertal av värdcellens proteiner, bland annat tillväxthämmarproteinerna p53 och retinoblastoma. Båda dessa proteiner reglerar bland annat celldelning, epiteldifferentiering och apoptos (programmerad celldöd).
De kliniska förändringar som en HPV-infektion kan ge upphov till i munslemhinnan är bland annat papillom, kondylom, fokal epitelial hyperplasi och vårtor. HPV kan identifieras hos cirka 10 – 25 procent av de patienter som har cancer i munnen. Den vanligaste HPV-typen som associeras med muncancer är HPV16. I cancerceller förekommer HPV oftast integrerad i värdcellens genom och kan då blockera virusets syntes av E2-protein som normalt kontrollerar avläsningen av HPV-cancergenerna.
De HPV-positiva formerna av oral cancer avviker i många avseenden från de HPV-negativa. De HPV-positiva formerna saknar i allmänhet mutationer i p53. Tumörprognosen är bättre än för de HPV-negativa formerna som kännetecknas av p53-mutationer. Om HPV-vacciner visar sig vara effektiva när det gäller att förebygga HPV-infektioner skulle man kunna förhindra upp till en femtedel av alla former av cancer i huvud-halsregionen.
Den rådande uppfattningen är att cirka 15 procent av alla former av cancer hos människan orsakas av humant papillomvirus. HPV är den viktigaste enskilda riskfaktorn för cancer i livmoderhalsen (1,2). Viruset anses spridas veneriskt och förekommer faktiskt mest bland yngre personer. Å andra sidan förekommer HPV också i munnen hos vuxna och barn samt vid cancer i huvud- och halsområdet. Det är därför uppenbart att viruset även måste kunna spridas på andra sätt (3 – 9). De förändringar som HPV orsakar i epitelceller som omvandlar dem till cancer är förhållandevis väl kända. Här ges en kort översikt över viruset, dess proteiner samt hur dessa proteiner stör celldelningen och inverkar på många av de värdcellsproteiner som styr cellens funktioner. De förändringar som HPV orsakar i munhålan beskrivs i korthet samt den aktuella uppfattningen om vilken betydelse HPV har för uppkomsten av muncancer – ett samband som vår forskningsgrupp föreslagit redan för 20 år sedan (10).
HPV:s genom
HPV är ett litet virus som har ett cirkulärt genom som består av dubbelsträngat DNA med en storlek på cirka 8 000 baspar. Genomet kan delas in i så kallade tidiga (early, E) och sena (late, L) läsramar (open reading frames, ORF) efter hur de bidrar till infektioner som leder till virusreplikation (Fig. 1). Läsramarna som kodar för proteiner finns endast i den ena DNA-strängen (2,11). De tidiga generna (E1–E7) uttrycks strax efter att infektionen skett, innan DNA: t börjar replikera (12,13). De proteiner som produceras reglerar replikationen och uttrycket av virusets DNA. Proteiner som produceras av virusens tidiga genregioner (i första hand E6 och E7) bidrar också till att göra värdcellen odödlig och omvandlar den till en malign cell (1,2,14). De sena generna (L1 och L2) ger däremot upphov till strukturella virusproteiner och aktiveras i slutskedet av viruscykeln (15). Den region i genomet som blir avläst motsvarar inte direkt ett visst protein. Ett proteins struktur kan nämligen bestämmas av flera regioner som fogas ihop. I Tabel 1 ses läsramarna för HPV och lokalisationen samt funktionen för de proteiner som läsramarna producerar.
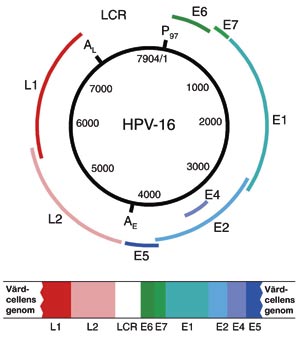
Fig. 1. Genomet för HPV typ 16 och integreringen av HPV.
Protein |
Storlek |
Lokalisation och mängd i cellen |
Funktion |
|---|---|---|---|
Sena |
|||
L1 |
55 – 60 kDA |
Cellkärna/++++ |
Viktigaste ytprotein i viruset (cirka 80 %) |
L2 |
70 kDA |
Cellkärna/++ |
Ytprotein |
Tidiga |
|||
E1 |
68—75 kDA600—650 aminosyror |
Cellkärna/+ |
Nödvändig för genomreplikering, aktiverar helikas och bibehåller virusgenomet episomalt, viktig transkriptionsfaktor |
E2 |
50 kDA400 aminosyror |
Cellkärna/+ |
Transkriptionsfaktor, deltar i replikation av virusgenomet |
E4 |
17 kDA: mest i form av fusionsprotein E1—E4, också formen 16—10 kDA förekommer |
Cytoplasma/+++ |
Packning av virusetViruspartikelmognadUpplösning av cytokeratinfilament |
E5 |
8—10 kDA |
Cytoplasma/+ |
Har interaktion med EGF-receptorn och aktiverar PDGF-receptorn, transformerande protein |
E6 |
16—18 kDA150—160 aminosyror |
Cellkärna/+ |
Förhindrar celldifferentiering, fragmenterar p53 i närvaro av E6-AP protein, transformerande protein i samverkan med RAS-onkogenen (se Tabell 2) |
E7 |
11 kDA100 aminosyror |
Cellkärna/++ |
Binder bland annat till pRB-105, P107 och P130, samverkan med RAS-onkogenen, transformerande protein, (se Tabell 3). |
HPV-genomet innehåller förutom läsramar också en lång kontrollregion, en region som inte producerar protein (long control region, LCR) (Fig. 1). Kontrollregionen är cirka 1 000 baspar stor. Många av värdcellens proteiner fäster vid denna region som viruset utnyttjar för att styra replikation av sitt genom och för cellens maligna omvandling (2,12,16). Även många av virusets egna proteiner (bland annat E2) fäster vid den här kontrollregionen och reglerar därmed bland annat uttrycket av cancergenerna E6 och E7. Virusets genom kan integreras med värdcellen i kromosomer. När detta sker öppnar sig den dubbelsträngade HPV-cirkeln (vanligen inom läsregionen E2) och viruset integreras slumpmässigt med värdcellens genom (Fig. 1, nedre delen). Integreringen leder till att E2-proteinets reglerande funktion på uttrycket av cancergenerna E6 och E7 hindras och möjliggör ett överuttryck av cancergenerna (16,17).
Klassifikation
Papillomvirusen är uråldriga och genomet har förändrats mycket lite under de senaste årmiljonerna. Virusen är artspecifika, det vill säga människans papillomvirus kan endast infektera människan och papillomvirus hos djur (som förekommer bland annat hos katt, nöt, hund och ren) kan endast infektera sin specifika art.
Ursprungligen klassificerades papillom- och polyomvirusen under samma grupp (polyomavirus) eftersom de föreföll lika när man studerade dem med elektronmikroskopi. (De var ikosahedrala DNA-virus med två strängar och saknade hölje.) Det framkom dock på 1980-talet att virustyperna trots allt har rätt lite gemensamt både vad gäller genomet och genfunktionerna.
För närvarande känner vi till 106 olika typer av papillomvirus hos människan men man uppskattar att det totala antalet HPV överskrider 200 (18). HPV-virusen kan grupperas efter den målvävnad de infekterar: slemhinnor (till exempel HPV 6, 11, 42) eller hudvävnad (HPV 2,4,7). Man kan också dela in HPV efter cancerrisk, det vill säga i lågrisktyper (HPV 6,11,42) och högrisktyper (HPV 16,18,31,33,35). Numreringen följer kronologin för när de upptäckts medan typningen baserar sig på bassekvensen i generna L1, E6 och E7. Om genomets bassekvens avviker med mer än 10 procent från en tidigare identifierad virustyp är det fråga om en ny typ. Om differensen är mindre än 10 – 2 procent är det en subtyp, och om sekvensdifferensen är mindre än 2 procent handlar det om en HPV-variant.
År 2004 ändrades klassifikationen av HPV-virusen i och med att de äntligen godkändes som en separat grupp (Papillomviridae) i den internationella virusklassifikationen (International Council of Taxonomy of Viruses, ICTV) (18). Den nya klassifikationen bygger på ett släktträd som för papillomvirusgenomet. Släktträdet består av olika släkten som i sin tur består av arter. Papillomvirusens släktträd innehåller 16 släkten som anges med grekiska bokstäver. De kliniskt mest betydelsefulla HPV-typerna hänförs till släktet a-papillomvirus som i sin tur delas in i arter. Bland arterna finns även papillomvirus som förekommer hos apa och schimpans (18). Huvuddelen av alla HPV-typer som associeras med skivepitelcellscancer hör antingen till art nummer 9 (HPV-typer 16, 31, 33, 35, 52, 58, 67) eller 7 (HPV 18, 39, 45, 59, 68, 70). HPV-typerna 2, 27 och 57 (som faller under art 4) orsakar vårtor och förekommer i munhålan. Under art 10 räknas HPV typ 6 och 11 som orsakar papillomen samt HPV 13 som orsakar fokal epitelial hyperplasi (FEH). FEH orsakas också av HPV 32 som tillhör art 1. Människans papillomvirus typ 13 anses ursprungligen ha varit ett schimpanspapillomvirus som genomgått mutationer för cirka fem miljoner år sedan.
Diagnostik
HPV går inte att odla in vitro och den serologiska diagnostiken är ännu i sin linda. Därför baserar sig HPV-diagnostiken på att visa förekomsten av virusets DNA eller RNA med olika molekylärbiologiska metoder. De genetiska variationerna mellan olika HPV-typer är minst på L1-området. Därför används detta område av virusgenomet som målsekvens vid DNA-diagnostik som baserar sig på genamplifiering (1). Det finns ett flertal kommersiella HPV-test, bland annat Hybrid capture 2 (Digene, Gaithersburg, Maryland, USA), Amplicor (Roche, Basel, Schweiz), PreTect HPV-Proofer (NorChip, Klokkarstua, Norge) och InnoLiPA (Innogenetics, Gent, Belgien). HPV-proteinet L1 kan också påvisas ur vävnadsprover med histoimmunologisk teknik men sådana prov visar endast positivt resultat om viruset produceras aktivt. Vid denna testning används i allmänhet en antikropp som tagits fram mot papillomvirus hos nöt och som även reagerar med många HPV-typer hos människa.
HPV-proteiner
Tabell 1 visar de proteiner som produceras av HPV-virusets läsramar, deras uttryck i cellen samt proteinernas centrala funktioner. E1 öppnar DNA-strängen och är nödvändig för replikationen av genomet samt för transkriptionsregleringen. E2 är en transkriptionsfaktor som också reglerar transkriptionen av E6 och E7. E4 binder vid cytoplasmats cellskelett. Nyligen har man visat att den även påverkar generna E6, E7 och L2. E5, E6 och E7 är transformerade proteiner vars funktion kommer att beskrivas detaljerat längre fram i artikeln.
HPV-högrisktyperna 16, 18, 26, 31, 33, 35, 39, 45, 52, 53, 56, 59, 66, 68, 73 och 82 ger cellen odödlighet och vissa transformeringsegenskaper. Detta beror främst på att virusets E6- och E7-gener producerar ämnen som kan binda ett stort antal av de proteiner som värdcellen producerat (Tabell 2 och 3) (19 – 59). Dessa proteiners funktion är bland annat reparation av DNA och reglering av celldelningen. De viktigaste målen för E6- och E7-proteinerna är «väktaren av människans genom» p53 samt retinoblastoma proteinet pBR – två centrala reglerare av celldelningscykeln. Särskilt HPV (med hög risk för att bli elakartad) har förmåga att tvinga cellen till delning eftersom viruset behöver proteiner som syntetiseras av värdcellen då den delar sig för sin egen replikation. Dessutom fördröjer HPV celldifferentieringen, hämmar reparationsmekanismerna för DNA, förhindrar den programmerade celldöden (apoptos) och gör den infekterade cellen osynlig för kroppens försvarsmekanismer. Dessa processer gör så småningom cellen odödlig. I slutet av detta invecklade förlopp kan cellen utvecklas till en cancercell eftersom HPV gör det möjligt för mutationer att lagras i värdcellens genom (16, 60 – 62). HPV-infektionen leder till förändringar på cell- och molekylärnivå som i dag är förhållandevis väl utredda.
Värdcellens protein |
Proteinets funktion vid bindning till värdcellen |
Litteraturreferens |
|---|---|---|
AMF-1/Gps2 |
Ökad transkription av p300 |
Degenhardt och Silverstein, 2001 (19) |
Bak |
Hör till Bcl-2-familjen, proapoptotiskt protein |
Du et al., 2004 (20) |
CBP/p300 |
Koaktivator med p53, reglerar signalöverföringen (aktiverar gener som ökar cellcykelreglering, celldifferentiering och försvarsrespons) |
Bernat et al., 2003 (21) |
c-myc |
Transkriptionsfaktor, inducerar apoptos |
Crook et al., 1988 (22) |
E6AP |
Reglerar signaleringen för cellproliferation genom att nedbryta Blk-kinas (hör till scr-familjen) |
Kehmeier et al., 2002 (23) |
E6TPI |
GAP-protein (GTPas-aktiverande protein), reglerar Rab-genen genom negativ återkoppling |
Singh et al., 2003 (24) |
ERC55(E6BP) |
Kalciumbindande protein (är av betydelse för epiteldifferentiering och för förhindrande av apoptos) |
Sherman et al., 2002 (25) |
hDLG/Sap97 |
Humangenen som motsvaras av DLG tillväxthämmargenen hos bananfluga. Är av betydelse för polariseringen* av epitelceller och uppkomsten av intercellulära bindningar |
Massimi et al., 2004 (26) |
hScrib |
Förekommer i de intercellulära bindningarna mellan epitelceller, motsvarar den tillväxthämmande genen Scrib hos bananflugan (reglerar intercellulära bindningar och hämmar epiteltillväxt). |
Nakagawa och Huibregtse, 2000 (27) |
Interferonreglerande faktor 3 (IRF3) |
Induktion av interferon; transaktivator av interferoner |
Ronco et al., 1998 (28) |
MAGI-1/2/3 |
Tight junctionproteiner; bildar komplex med b-katein, reglerar PTEN-genen (tillväxthämmare) |
Grm och Banks, 2004 (29) |
Mcm7 |
Gen som gör DNA-replikation möjlig och initierar den |
Brake et al., 2003 (30) |
Mupp1 |
Polymorft PDZ-protein; är av betydelse i signalöverföringen |
Lee et al., 2000 (31) |
Paxillin |
Lokalt adhesionsprotein, är av betydelse för celladhesionen och för reglering av aktinet i cytoskeleton |
Tong och Howley, 1997 (32) |
p53 |
Tillväxthämmargen; reglerar cellresponsen till mitogena signaler |
Scheffner et al., 1990 (33) |
XRCC1 |
DNA-reparationsprotein |
Iftner et al., 2002 (34) |
Värdcellens protein |
Proteinets funktion vid bindning till värdcellen |
Litteraturreferens |
|---|---|---|
pRB, p107, p130 |
Transkriptionsfaktor och mitosreglering |
Dyson et al., 1992 (35) Gonzales et al., 2001 (36)Wu et al., 1993 (37) |
p27 |
Hämmare av cyklinberoende kinas |
Zerfass-Thome et al. 1996 (38) |
p21 |
Hämmare av cyklinberoende kinas |
Funk et al., 1997, (39) Jones et al., 1997a (40) |
TBP |
Medverkar centralt vid initiering av transkriptionen, protein som binder sig vid TATA-boxen |
Massimi et al., 1996 (41) |
TAF110 |
Medverkar centralt vid initiering av transkriptionen, protein som binder sig vid TATA-boxen, transkriptionsreglerare |
Mazzarelli et al., 1995 (42) |
AP-1 |
Transkriptionsfaktor |
Antinore et al., 1996 (43) |
Mi2 (HDAC) |
Histondeacetylas |
Brehm et al., 1999a (44) |
IGFBP-3 |
Mål för transkription av p53, reglerar den intracellulära mängden av insulinliknande tillväxtfaktor |
Mannhardt et al., 2000 (45) |
M2 pyruvatkinas |
Reglerar det glykolysassocierade enzymet M2 |
Zwerschke et al., 1999 (46) |
Glukosidas |
Glykolysreglerande enzym |
Zwerschke et al., 2000 (47) |
hTid-1 |
Humangenen som motsvaras av tillväxthämmargenen Tid56 dnaJ, reglerar apoptos |
Schilling et al., 1998 (48) |
p48 (ISGF3) |
Interferonreglerande protein |
Barnard och McMillan, 1999 (49) |
IRF-1 |
Reglerar IFN-g |
Park et al. 2000 (50) |
Aktin F |
Rey et al., 2000 (51) |
|
MPP2 |
Transkriptionsfaktor |
Lüscher-Firzlaff et al., 1999 (52) |
S4 ATPas |
Del S4 av protosomet 26S |
Berezutskaya och Bagchi, 1997 (53) |
Oct-4 |
Transkriptionsfaktor |
Brehm et al., 1999b (54) |
Skip |
Protein som binder sig vid onkogenen Ski, reglerar responsen till TGF (transformerande tillväktfaktor) |
Prathapam et al., 2001 (55) |
E2F-1 |
Transkriptionsfaktor |
Hwang et al., 2002 (56) |
Kinas B1 |
Reglerar cellcykeln vid G2-fasen |
Spitkovsky et al., 2002 (57) |
Smad2, 3, 4 |
Reglerar TGF-signaltransduktionen |
Lee et al., 2002 (58) |
P/CAF |
Aktivering av IL8-promotorn i samverkan med CBP/p300 |
Huan et al., 2002 (59) |
Viruscykeln
Det sannolika målet för papillomvirusinfektionerna är skivepitelvävnadens basalceller (11, 64, 65). HPV utnyttjar receptorer på målcellens yta för att infektera cellen. Receptorerna på HPV är ännu inte helt kända men heparansufat och a6b4-integrin är goda receptorkandidater (65, 66). Efter infektionen binder värdcellens proteiner till LCR-regionen och inleder transkriptionen av E1- och E2-generna i HPV-viruset.
E6- och E7-generna stör cellens normala delning på många olika sätt. E6 binder till proteinet som «genomväktargenen» p53 framkallar, medan E7 bland annat binder till retinoblastomproteinet (pRb). Därefter läses generna E1 och E2 av. E2 hindrar avläsningen av E6 och E7 och gör det möjligt för E1 att fästa vid den plats inom regleringsområdet som inleder replikeringen av virusets genom. I epitelets mellanskikt uttrycks nu både E4 och E5 och inom epitelets ytskikt uttrycks de sena generna L1 och L2 och gör det möjligt för mogna viruspartiklar att produceras (11, 67, 68) (Fig. 2).
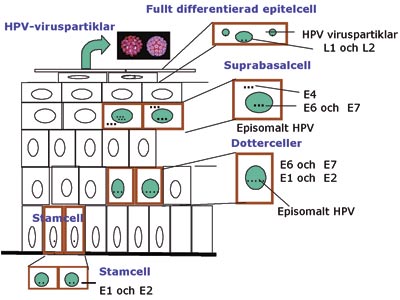
Fig. 2. Uttrycket av HPV-proteinerna i epitelceller är beroende av epitelets differentieringsgrad.
HPV stör cellcykeln
Ett flertal nya rön har visat att de proteiner som syntetiserats genom uttryck av de tidiga HPV-generna kan leda till att kontrollpunkterna i cellcykeln ignoreras genom att de binder till cyklin och cyklinassocierade kinaskomplex eller andra faktorer som reglerar cellcykeln. De bäst kända av dessa proteiner är de som uppkommer genom uttryck av cancergenerna E6 och E7 av HPV-högrisktyper som binder till många proteiner som är centrala för cellcykeln (19 – 59) (Tabell 2 och 3). De viktigaste proteinerna av detta slag är p53 som E6 binder till samt retinoblastomproteinet som E7 binder till (33 – 37). HPV-proteinet E5 stör också cellcykeln på många olika sätt och verkar som transformerande protein (13, 16).
På grund av HPV-infektionen kan cellen bli odödlig och omvandlas till en cancercell. HPV-genomet kan enbart replikeras när värdcellen delar sig. Därför tvingar de HPV-relaterade proteinerna värdcellen till celldelning. Celldifferentieringen fördröjs och epitelvävnadens ytskikt är kraftigt differentierat för att produktionen av viruspartiklar ska kunna sättas igång. När cellen delar sig utsätts genomet för mutationer som under normala förhållanden korrigeras av cellens genomväktare, p53. Men när cellen infekterats av HPV kan p53 inte längre identifiera de fel som uppstår. Cellen delar sig och mutationerna anhopas i genomet. Som bieffekt bildas en odödlig cell som småningom blir elakartad.
För att cellcykeln ska kunna gå vidare mot mitos sker olika kontroller inom cellen: 1) Har cellen rätt storlek inför mitosen, 2) är DNA oskadat samt 3) är miljön i cellen gynnsam? Vid GI-stadiet spjälkas cykliner och det bildas hämmande proteiner som i sin tur hämmar de cyklinberoende kinaserna (cdk 1 – 9). De cyklinberoende kinaserna förs in i sitt målprotein genom att de kopplar på en fosfatgrupp till målproteinets serin- eller treoninaminosyra. Initialt under HPV-infektionen fungerar G2 (en av cellcykelns övriga viktiga kontrollpunkter) men till slut leder mutationerna i värdcellens genom (som delvis beror på inverkan av E6) till en funktionsstörning i cellcykelns G2-kontrollpunkt. HPV 16 kan påverka G2/M-kontrollpunkten på många olika sätt, exempelvis genom att öka uttrycket av cdk2, cyklin A och cyklin B (2, 16, 58).
Cellcykeln och HPV E7
Cykliner är proteiner som uttrycks i cellen vid givna faser i cellcykeln. Dessa proteiner reglerar cellcykeln genom att binda till de cyklinberoende kinaserna och aktivera dem. Fig. 3 visar de faser i cellcykeln då de enskilda cellcyklinerna uttrycks samt hur E7-proteinet från högrisk-HPV stör så väl funktionen av cyklinerna, de cyklinberoende kinaserna som cdk-hämmarna (58). E7-proteinet gör det lättare att passera den sena kontrollpunkten G2/M i cellcykeln och leder till att replikationen av kromosomerna blir ofullständig. Det uppstår anaeuploida celler, celler där kromosomantalet avviker från det normala (Tabell 3) (35 – 59).
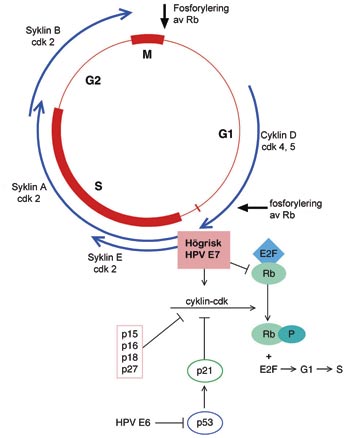
Fig. 3 . Cancerproteinet E7 från högrisk-HPV binder sig vid retinoblastomproteinet, (pRb), vilket frisätter transkriptionsfaktor E2F och för cellcykeln vidare.
Cellcykeln och E6
E6 från högriskpapillomviruset tar sig förbi G1 i ett tidigt skede och bryter ner p53 sedan p53 kopplats till E6 (23,33). Nedbrytningen beror på att proteinet E6AP binds till E6. Att E6AP binds till E6 är en nödvändig förutsättning för E6 ska kunna binda till p53. När p53 bryts ner hämmas funktionen av den cyklinberoende kinashämmaren p21 vilket i sin tur gör det lättare för viruset att passera kontrollen vid G1 (Fig. 4).
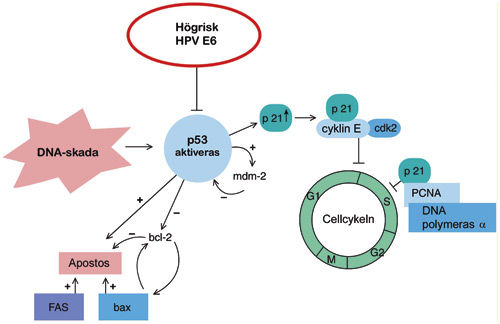
Fig. 4.Värdcellens p53 fragmenteras under inverkan av E6-proteinet från högrisk-HPV; cellcykeln fortsätter från G1-fasen direkt till syntesfasen (S).
Kliniska fynd i munnen associerade till HPV-infektioner
HPV-infektioner ger upphov till godartade tumörer i munnen: vårtor, papillom/kondylom och fokal epitelial hyperplasi. Man har också påträffat papillomvirus i orala lichenläsioner, leukoplakier och erytroplakier. Virusets roll vid uppkomsten av dessa tillstånd är dock fortfarande oklar (1,69). Tidigare ansåg man att HPV smittar via oralt könsumgänge. Våra tidiga undersökningar visar dock att barn kan vara infekterade i munnen eller könsorganen med HPV. Barnen har uppenbarligen infekterats av modern redan under förlossningen. Hos vuxna medför kronisk oral HPV-infektion att partnerns risk för att få oral HPV-infektion ökar tiofaldigt (70 – 72). Det naturliga förloppet hos de orala infektionerna påminner om det naturliga förloppet hos HPV-infektioner i könsorganen (72). Cirka 10 procent av populationen har HPV-DNA i vad som ser ut att vara en frisk munslemhinna. Tandköttsfickorna och tonsillerna kan vara dolda infektionshärdar för HPV-infektioner i munhålan (73,74).
Orala vårtor uppstår oftast som en följd av infektion med HPV-typerna 2, 4 och 57. Den kliniska bilden är både varierande och ospecifik. Baserat på kliniska fynd går det dock inte att avgöra om det gäller en vårta av den typ som uppstår på huden eller slemhinnepapillom respektive slemhinnekondylom. Papillom är godartade, blomkålsformade epiteliala tumörer som oftast orsakas av HPV 6 eller 11 (1,69). Hos barn utgör papillomen 7 – 8 procent av samtliga godartade tumörer jämfört med 0,5 – 3,0 procent i den vuxna populationen. Tidigare ansågs kondylom och papillom vara två olika tillstånd både etiologiskt och histologiskt. I takt med att DNA-tekniken utvecklats har man kunnat visa att såväl kondylomen som papillomen snarast är relaterade till infektion med HPV 6 och/eller HPV 11. Det är dock tänkbart att en del av papillomen kan uppstå på grund av mekanisk irritation (1, 69).
I litteraturen beskrevs fokala epiteliala tumörer (FEH) första gången redan på 1800-talet. Som namnet antyder består FEH av små, ljusa upphöjningar i munslemhinnan. Förändringarna kan förekomma isolerat eller, vilket är vanligare, i form av multiforma förändringar som ses i underläppen och på andra håll i munhålans slemhinna (1,69). Det finns också karaktäristiska FEH-celler som troligen är celler i apoptos. FEH är ett godartat tillstånd som orsakas av HPV typ 13 och 32 som endast förekommer i munnen. Det har dock nyligen beskrivits två fall där FEH övergått i cancer hos två patienter som behandlats med bromsmedicin mot HIV (HART). FEH är ärftligt: HLA-DRB10404-allelen associeras med en ökad risk för kliniska infektioner som beror på HPV 13 (75).
Muncancer och HPV
År 2002 var den globala incidensen av oral cancer 274 850. I utvecklingsländerna låg oral cancer på sjunde plats bland de vanligaste maligna sjukdomarna hos män och på åttonde plats bland kvinnor (76) jämfört med i-länderna 11 för män och 15 för kvinnor. År 2002 avled 127 783 människor i den här formen av cancer (76). De mest betydelsefulla riskfaktorerna för muncancer är rökning och alkoholkonsumtion. Risken är dosrelaterad: Ökad konsumtion av såväl tobak som alkohol ökar risken flerfaldigt. Riskfaktorerna förklarar 80 – 85 procent av alla fall av muncancer.
En ny metaanalys (77) visar att sannolikheten för att identifiera HPV-DNA i munslemhinnan växer i takt med graden av dysplasi. I 94 undersökningar analyserades 4 680 prover och forskarna uppgav att sannolikheten för att upptäcka HPV i frisk munslemhinna var 10 procent (95 % CI = 6,1 – 14,6), i leukoplakiförändringar 22 procent (95 % CI = 15,7 – 29,9), verruköst carcinom 29,5 procent (95 % CI = 23,0 – 36,8) och skivepitelcellscancer 46,5 procent (95 % CI = 37,5 – 55,5). Sannolikheten för att finna en högriskvariant av HPV var 2,8 gånger högre än att finna en lågriskvariant (0,24; 95 % CI=0,16 – 0,33 respektive 0,09; 95 % CI = 0,06 – 0,13).
I litteraturen har man fram till slutet av år 2004 beskrivit 6 500 fall av oral cancer där man använt HPV-testning. Av dessa var 20 procent positiva (5,6). Den vanligaste HPV-typen var typ 16 (cirka 80 % av fallen). Näst vanligast var typ 18. I några fall av muncancer har också lågriskvarianterna 6 och 11 påträffats. På senare tid har ett flertal så kallade case-controlstudier som fastställer HPV-infektionen som en självständig riskfaktor för oral cancer publicerats. Av dessa framgår det att HPV-infektion ökar risken för cancer 5,4 gånger (78 – 80). Forskningen visar att särskilt cancer i tunga och tonsiller har samband med HPV-infektion. Patienter med HPV-positiv oral cancer tycks ha en bättre prognos än patienter med HPV-negativ cancer. HPV-infektioner förekommer oftare hos icke-rökare än hos rökande muncancerpatienter. Enligt en nordisk undersökning kunde HPV L1-antikroppar påvisas två gånger oftare hos personer som fick tungcancer efter 10 – 20 år jämfört med kontrollpersoner (81). Även detta fynd talar för att exponering för HPV föregår sjukdomsutbrott.
Virusrelaterade särdrag och muncancer
Inte bara HPV-typen är relaterad till uppkomsten av cancerceller. Andra viktiga virusrelaterade faktorer är; 1) det totala antalet virus i den infekterade cellen, 2) virusets integrering i värdcellens genom samt 3) hur stort uttrycket av E6- och E7-cancergener är. Integreringen av viruset i värdcellens genom kan bland annat undersökas med hjälp av tvådimensionell så kallad Southern blot hybridiseringsteknik eller PCR där det genområde som förstärks utgörs av HP-virusets E2. Denna gen bryts nämligen upp i mindre beståndsdelar vid integreringsprocessen och bortfaller delvis vilket ger ett negativt resultat vid PCR. Den nyaste metoden kallas realtids-PCR och betyder också att man kan bestämma mängden av den gen som förstärks eller dess mRNA. Metoden har använts för att identifiera integrerat HPV 16-material i prover (82). Koskinen et al. (83) kunde med hjälp av realtids-PCR identifiera HPV i 16 av 61 biopsier från cancer i mun, huvud och hals. Virusgenom fanns integrerat i 48 procent av fallen och episomalt i 35 procent. I 17 procent av muncancerbiopsierna fann man bägge formerna.
Antalet replikerade viruskopior i muncancervävnaden är ännu inte helt känt. Studier harvisat att endast en del av cancercellerna är positiva för virus och att virusmängden dessutom är låg i dessa celler. Det skulle kunna betyda att endast en del av HPV replikeras i cancercellerna alternativt att cancern inte är av klonalt ursprung. I sina studier fann Koskinen et al. (83) endast 1 – 97 viruskopior per 10 000 orala cancerceller. De senaste åren har man inte bara studerat förekomsten av virusgenom i cancerceller utan också uttrycket av dess cancergener för att om möjligt kunna belysa den virala aktiviteten (84 – 86). I cirka hälften av fallen av HPV-DNA-positiva muncancerformer var mRNA uttryckt. Detta uttryck var associerat med normalt p53 i tumören (wild-type) (86) med nedsatt pRB och ett överuttryck av p16 i tumörerna. Uttrycket av HPV 16 E6/E7 har också associerats med en specifik morfologi i cancervävnaden (87) och en nedsatt allelförlust hos 13 av 15 markörer för kromosomerna 3p, 9p och 17p (86).
Både terapeutiska och profylaktiska vacciner har utvecklats mot HPV. En del av dessa vacciner provas för närvarande i kliniska studier. Om de profylaktiska vaccinerna visar sig vara effektiva skulle upp till 25 procent av de olika cancerformerna i huvud- och halsområdet kunna förebyggas (88).
English summary
Syrjänen S.
Human papillomaviruses and oral infections
90 – 7.
Over the past 25 years, interest in human papillomaviruses (HPV) has increased remarkably, because of their potential role in pathogenesis of several human malignancies. Today, 106 HPV types have been identified of which almost 40 have been detected in oral mucosa.
In 1983, Stina Syrjänen and her group published the first evidence suggesting that HPV might be involved in pathogenesis of oral squamous cell carcinoma. The recognition of morphological similarities between oral and previously established genital HPV lesions led to this pioneering proposal. In the recent meta-analyses of the existing evidence as well as in several case-control studies, HPV has been confirmed as an independent risk factor for oral squamous cell carcinoma. The evidence indicates that HPV associated oral cancers form an entity with a better survival. HPV can also cause benign lesions in oral mucosa, such as papillomas, condylomas, warts and focal epithelial hyperplasia (FEH).
HPV is also detectable in healthy mucosa of adults and infants. HPV has been traditionally regarded as a sexually transmitted disease, but the recent evidence implicates the existence of other modes of transmission. It is now apparent that parts of the HPV infections in newborns are acquired vertically from the mother during delivery. Of the oncogenic HPV types, HPV16 is the most prevalent type in oral carcinoma. Viral copy numbers in oral cancers are generally low, suggesting a non-clonal association of the tumor with HPV. If this is the case, HPV is not needed for maintenance of the malignant state. A hit and run theory has been proposed several times to explain the development of HPV-negative tumors. Of the eight open reading frames (ORF), the early genes and particularly E6 and E7 are the most important oncogenes involved in HPV-induced immortalization and cell transformation. E6 and E7 oncoproteins are able to interfere with several cellular proteins playing key role in controlling cell cycle, apoptosis and cellular differentiation. “The guardian» of the cell genome, p53 binds with E6 in the presence of E6AP leading to the degradation of p53, and similarly E7 binds to retinoblastoma, pRB. In addition, the HPV E5 protein has some transforming features. HPV cannot be cultivated but replication of the virus is dependent on epithelial differentiation with an unknown mechanism. Currently, clinical trials are ongoing on prophylactic and therapeutic vaccines for HPV aiming to global eradication of cervical cancer. If these vaccinations are successful, there is a possibility that also up to 25 % of head and neck cancers can be prevented by these prophylactic HPV vaccines.
Litteratur
1. Syrjänen K, Syrjänen S. Papillomavirus infections in human disease. New York: Wiley; 2000. p. 1 – 615.
2. zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2002; 2: 342 – 50.
3. Syrjanen S, Puranen M. Human papillomavirus infections in children: the potential role of maternal transmission. Crit Rev Oral Biol Med 2000; 11: 259 – 74.
4. Syrjänen S. HPV infections in children. Invited review. Papillomvirus Report 14: 93 – 110, 2003.
5. Syrjanen S. Human papillomavirus (HPV) in head and neck cancer. J Clin Virol 2005; 32S: S59–S66.
6. Syrjänen S. Human papillomavirus (HPV) in oral cancer. J Clin Pathol (In press).
7. Snijders PJ, Steenbergen RD, Meijer CJ, Walboomers JM. Role of human papillomaviruses in cancer of the respiratory and upper digestive tract. Clin Dermatol 1997; 15: 415 – 25.
8. Smith EM, Hoffman HT, Summersgill KS, et al. Human papillomavirus and risk of oral cancer. Laryngoscope 1998; 108: 1098 – 103.
9. Herrero R. Human papillomavirus and cancer of the upper aerodigestive tract. J Natl Cancer Inst Monogr 2003; 31: 47 – 51 [Chapter 7].
10. Syrjänen K, Syrjänen S, Lamberg M, et al. Morphological and immunohistochemical evidence suggesting human papillomavirus (HPV) involvement in oral squamous cell carcinogenesis. Int J Oral Surg 1983; 12: 18 – 24.
11. Doorbar J. The papillomavirus life cycle. J Clin Virol 2005; 32S: S7–S15.
12. Chow LT, Broker TR. Papillomavirus DNA replication. Intervirol 1994; 37: 150 – 8.
13. Fehrmann F, Klumpp DJ, Laimins LA. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J Virol 2003; 77: 2819 – 31.
14. Dürst M, Dzarlieva-Petrusevka T, Boukamp P, Fusenig NE, Gissmann L. Molecular and cytogenetic analysis of immortalized human primary keratinocytes obtained after transfection with human papillomavirus type 16 DNA. Oncogene 1987; 1: 251 – 6.
15. Florin L, Sapp C, Streeck RE, Sapp M. Assembly and translocation of papillomavirus capsid proteins. J Virol 2002; 76: 1009 – 14.
16. Munger K, Howley PM. Human papillomavirus immortalization and transformation functions. Virus Res 2002; 89: 213 – 28.
17. Wentzensen N, Vinokurova S, von Knebel DM. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res 2004; 64: 3878 – 84.
18. de Villiers EM, Fauquet C, Broker TR, Berhard HU, zur Hausen H. Classification of papillomaviruses. Virology 2004; 324: 17 – 27.
19. Degenhardt YY, Silverstein SJ. Gps2, a protein partner for human papillomavirus E6 proteins. J Virol 2001; 75: 151 – 60.
60. Cottage A, Dowen S, Roberts I, Pett M, Coleman N, Stanley M. Early genetic events in HPV immortalized keratinocytes. Genes, Chromosomes Cancer 2001; 30: 72 – 9.
61. Duensing S, Munger K. Centrosome abnormalities and genomic instability induced by human papillomavirus oncoproteins. Prog Cell Cycle Res 2003; 5: 383 – 91.
62. Munger K, Basile JR, Duensing S, Eichten A, Gonzalex SL, Grace M, et at. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene 2001; 20: 7888 – 98.
63. Stanley MA, Browne HM, Appleby M, Minson ACIRKAProperties of a non-tumorigenic human cervical keratinocyte cell line. Int J Cancer 1989; 43: 672 – 6.
64. Egawa K. Do human papillomaviruses target epidermal stem cells? Dermatology 2003; 207: 251 – 4.
65. Culp TD, Christensen ND. Kinetics of in vitro adsorption and entry of papillomavirus virions. Virology 2004; 319: 152 – 61.
66. Giroglou T, Florin L, Schafer F, Streeck RE, Sapp M. Human papillomavirus infection requires cell surface heparin sulfate. J Virol 2001; 75: 1565 – 70.
67. Peh WL, Middleton K, Christensen N, Nicholls P, Egawa K, Sotlar K, et al. Life cycle heterogeneity in animal models of human papillomavirus-associated disease. J Virol 2002; 76: 10401 – 16.
68. Sherman L, Itzhaki H, Jackman A, Chen JJ, Koval D, Schlegel R. Inhibition of serum- and calcium-induced terminal differentiation of human keratinocytes by HPV 16 E6: study of the association with p53 degradation, inhibition of p53 transactivation, and binding to E6BP. Virology 2002; 292: 309 – 20.
69. Syrjänen S. Human papillomavirus infections and oral tumors. Med Microbiol Immunol (Berl) 2003; 192: 123 – 8.
70. Puranen M, Yliskoski M, Saarikoski S, Syrjanen K, Syrjanen S. Vertical transmission of human papillomavirus from infected mothers to their newborn babies and persistence of the virus in childhood. Am J Obstet Gynecol 1996; 174: 694 – 9.
71. Rintala M, Grénman S, Puranen M, Isolauri E, Ekblad U, Kero P, et al. Transmission of high-risk human papillomavirus (HPV) between parents and infant: a prospective study of HPV in families in Finland. J Clin Microbiol 2005; 43: 376 – 81.
72. Rintala M, Grénman S, Syrjänen S. Natural history of oral human papillomasvirus infections in female and male partners: a prospective follow-up study. J Clin Virol (In press).
73. Hormia M, Willberg J, Ruokonen H, Syrjanen S. Marginal periodontium as a potential reservoir of human papillomavirus in oral mucosa. J Periodontol 2005; 76: 358 – 63.
74. Syrjänen S. HPV infections and tonsillar carcinoma. J Clin Pathol 2004; 57: 449 – 55.
75. Garcia-Corona C, Vega-Memije E, Mosqueda-Taylor A, Yamamoto-Furusho JK, Rodriguez-Carreon AA, Ruiz-Morales JA, et al. Association of HLA-DR4 (DRB1*0404) with human papillomavirus infection in patients with focal epithelial hyperplasia. Arch Dermatol 2004; 140: 1227 – 31.
76. Ferlay J, Bray P, Pisani P, Parkin DM. Globocan 2002. Cancer incidence, mortality and prevalence worldwide. IARC CancerBase No.5, version 2.0. Lyon: IARCPress; 2004.
77. Miller CS, Johnstone B. Human papillomavirus as a risk factor for oral squamous cell carcinoma: a meta-analysis, 1982 – 1997. Oral Surg Oral Med Oral Radiol Endod 2001; 91: 622 – 35.
78. Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst 2000; 92: 709 – 20.
79. Herrero R, Castellsague X, Pawlita M, et al. Human papillomavirus and oral cancer: the International Agency for Research on Cancer Multicenter Study. J Natl Cancer Inst 2003; 95: 1172 – 83.
80. Schwartz SM, Daling JR, Doody DR, et al. Oral cancer risk in relation to sexual history and evidence of human papillomavirus infection. J Natl Cancer Inst 1998; 90: 1626 – 36.
81. Mork J, Lie AK, Glattre E, et al. Human papillomavirus infection as a risk factor for squamous-cell carcinoma of the head and neck. N Engl J Med 2001; 344: 1125 – 31.
82. Peitsaro P, Johansson B, Syrjänen S. Integrated human papillomavirus type 16 is frequently found in cervical cancer precursors as demonstrated by a novel quantitative real-time PCR technique. J Clin Microbiol 2002; 40: 886 – 91.
83. Koskinen WJ, Chen RW, Leivo I, Makitie A, Back L, Kontio R, et al. Prevalence and physical status of human papillomavirus in squamous cell carcinomas of the head and neck. Int J Cancer 2003; 107: 401 – 6.
84. Wiest T, Schwarz E, Enders C, Flechtenmacher C, Bosch FX. Involvement of intact HPV16 E6/E7 gene expression in head and neck cancers with unaltered p53 status and perturbed pRb cell cycle control. Oncogene 2002; 21: 1510 – 7.
85. Balz V, Scheckenbach K, Gotte K, Bockmuhl U, Petersen I, Bier H. Is the p53 inactivation frequency in squamous cell carcinomas of the head and neck underestimated? Analysis of p53 exons 2 – 11 and human papillomavirus 16/18 E6 transcripts in 123 unselected tumor specimens. Cancer Res 2003; 63: 1188 – 91. Erratum in: Cancer Res 2004; 64: 220.
86. Braakhuis BJ, Snijders PJ, Keune WJ, Meijer CJ, Ruijter-Schippers HJ, Leemans CR, et al. Genetic patterns in head and neck cancers that contain or lack transcriptionally active human papillomavirus. J Natl Cancer Inst. 2004; 96: 998 – 1006.
87. van Houten VM, Snijders PJ, van den Brekel MW, Kummer JA, Meijer CJ, van Leeuwen B, et al. Biological evidence that human papillomaviruses are etiologically involved in a subgroup of head and neck squamous cell carcinomas. Int J Cancer 2001; 93: 232 – 5.
88. Frazer IH. Prevention of cervical cancer through papillomavirus vaccination. Nat Rev Immunol. 2004; 4: 46 – 54.
Korrespondens: Professor Stina Syrjänen, Avdelningen för oralpatologi och oralradiologi, Odontologiska institutionen, Medicinska fakulteten, Åbo universitet, Lemminkäinengatan 2, 20520 ÅBO, Finland. E-post: stina.syrjanen@utu.fi
Referensgranskad artikel.