Sykdommer som affiserer hud og munnslimhinne
Forfattere
Hudavdelingen, Haukeland Universitetssjukehus, Bergen
Odontologisk institutt – oral patologi og rettsodontologi, Det odontologiske fakultet, Universitetet i Bergen
Artikkelen publiseres parallelt i Tidsskrift for Den norske lægeforening nr. 9, 2006
Bakgrunn. En rekke sykdommer kan gi forandringer i både hud og munnslimhinne.
Materiale og metode. Denne artikkelen, som er basert på aktuell litteratur og egne kliniske erfaringer, gir en oversikt over arvelige, autoimmune og andre inflammatoriske tilstander som kan affisere både hud og munnslimhinne.
Resultater. Av de aktuelle sykdommene forekommer lichen planus, erythema multiforme og lupus erythematosus hyppigst og har oftest god prognose. De blemmedannende sykdommene bulløst pemfigoid og pemfigus er sjeldne og medfører betydelig morbiditet, noe som ofte krever aggressiv immunsuppressiv behandling. Stevens-Johnsons syndrom og toksisk epidermal nekrolyse er potensielt livstruende tilstander. Det samme gjelder alvorlige former av epidermolysis bullosa, som er en gruppe genetisk betingede sykdommer med ulik arvegang og klinisk uttrykksform.
Fortolkning. Leger og tannleger bør ha kjennskap til kliniske presentasjonsformer og muligheter for diagnostikk av disse sykdommene. Histologisk vurdering og immunfluorescensundersøkelse er viktige supplementer til klinisk undersøkelse.
Både hud og munnslimhinne er kledd av flerlaget plateepitel, og mange sykdommer kan manifestere seg i begge organer. Hudaffeksjonen kan forutgå slimhinneforandringene og vice versa. Vi gjennomgår i denne artikkelen noen sentrale hud- og slimhinnesykdommer, særlig genetiske, autoimmune og andre inflammatoriske tilstander, med vekt på klinisk presentasjon og diagnostikk.
Infeksiøse sykdommer og kreft omtales ikke. Artikkelen bygger i hovedsak på oversiktsartikler i sentrale tidsskrifter og lærebøker i munnhulesykdommer og dermatologi (1, 2), foruten egen klinisk erfaring.
Epidermolysis bullosa
Epidermolysis bullosa er en gruppe sjeldne, medfødte sykdommer som skyldes defekter i overgangssonen mellom epitel og bindevev. På basis av elektronmikroskopi, molekylærgenetiske undersøkelser og klinisk forløp inndeles disse sykdommene i hovedgruppene simplex, junctionalis og dystrophica (3). Både autosomalt dominant og autosomalt recessiv arvegang er beskrevet.
Epidermolysis bullosa kan være til stede ved fødselen eller debutere i tidlig barnealder. Kardinalsymptomet er residiverende bulladanning som følge av tangentielle traumer mot hud og slimhinner. Blemmene kan tilhele med eller uten arrdanning. Det kliniske forløpet varierer sterkt. Mens lettgradige tilfeller av simplexvarianten kun affiserer huden og ofte «brenner ut» i voksen alder, er andre typer av tilstanden preget av uttalt og vedvarende affeksjon av både hud og munnslimhinne. Tannemaljedefekter ses hyppig. Andre slimhinner, negler og hårfollikler kan også affiseres. Subtypen epidermolysis bullosa junctionalis Herlitz er letal i spedbarns- eller tidlig småbarnsalder. Epidermolysis bullosa dystrophica Hallopeau-Siemens gir utbredt affeksjon av slimhinner i munn, pharynx og oesophagus og kompliseres av strikturerende arrdanning. Tilstanden er også assosiert med sterkt økt risiko for plateepitelkarsinom i huden, hvilket ofte er dødelig hos disse pasientene.
Behandlingen av epidermolysis bullosa er symptomatisk. Tannbehandling og prosedyrer som krever intubasjonsanestesi bør foregå hos spesialister som har erfaring med tilstanden, oftest ved universitetsklinikkene. Genetisk veiledning er essensielt.
Andre arvelige tilstander som affiserer hud og munnslimhinne er svært sjeldne (Ramme 1).
Ramme 1
Arvelige sykdommer som kan affisere hud og munnslimhinne (acrodermatitis enteropatica finnes også i akkvirert form)
Epidermolysis bullosa-sykdommene
Pseudoxanthoma elasticum
Acrodermatitis enteropatica
Ektodermal dysplasi-sykdommer
Dyskeratosis follicularis
Dyskeratosis congenita
Ehlers-Danlos’ syndrom
Incontinentia pigmenti
Pachyonychia congenita
Autoimmune blemmedannende tilstander
Pemfigusgruppen
Pemfigussykdommene er autoimmune hud- og slimhinnesykdommer karakterisert ved intraepitelial blemmedanning som følge av at kontakten mellom epitelcellene brytes ned (akantolyse). De overflatiske blemmene brister lett og etterlater seg smertefulle sårflater med fare for væsketap og elektrolyttforstyrrelser. Nikolskys tegn er positivt, dvs. at ikke-affisert perilesjonell hud løsner når man skyver på den. I munnslimhinnen finnes typisk utbredte og smertefulle erosjoner som tilheler langsomt, men uten arrdanning.
Funn ved histologiske prøver og immunfluorescensundersøkelse er typiske (Tabell 1), og det kan påvises sirkulerende antistoffer mot keratinocyttantigener. Basert på hvor i epitelet spaltingen skjer, inndeles sykdommen i pemphigus vulgaris, pemphigus vegetans, pemphigus foliaceus og pemphigus erythematosus. De to sistnevnte er rene hudsykdommer, mens pemphigus vegetans oppfattes som en sjelden variant av pemphigus vulgaris. Selv om pemphigus vulgaris er den hyppigst forekommende varianten, er tilstanden sjelden, med en årlig insidens på 0,1 – 0,5 tilfeller per 100 000 individer (4). Den rammer oftest personer mellom 40 og 60 år, men kan forekomme i alle aldersgrupper.
Sykdom |
Nedslag |
Lokalisasjon |
|---|---|---|
Pemfigus |
IgG og/eller C3, ev. IgA |
Intercellulært i epitel |
Paraneoplastisk pemfigus |
IgG og/eller C3 |
Intercellulært i epitel og i basalmembransonen |
Bulløst pemfigoid |
IgG og/eller C3 |
Basalmembransonen (lineært) |
Slimhinnepemfigoid |
IgG og/eller C3 |
Basalmembransonen (lineært) |
Dermatitis herpetiformis |
IgA, ev. IgM og/eller C3 |
Bindevevets papillspisser (granulært) |
Lineær IgA-dermatose |
IgA, ev. IgG, IgM og/eller C3 |
Basalmembransonen (lineært) |
Lichen planus |
Fibrinogen |
Basalmembransonen, strekker seg flammeformet inn i bindevevet |
Lupus erythematosus |
IgG, ev. IgM og C3 |
Basalmembransonen |
Hos et flertall av pasientene debuterer pemphigus vulgaris med munnhulelesjoner, hyppigst i kinnslimhinnen, mens hudforandringene gjerne tilkommer flere måneder senere (5, 6). Andre slimhinner, som i pharynx, larynx, oesophagus, conjunctiva og anogenitalt, affiseres i varierende grad. Tilstanden er alvorlig med høy morbiditet og var tidligere forbundet med svært høy mortalitet (70 – 90 %), sekundært til infeksjoner, ernæringsproblemer og væske-/elektrolyttforstyrrelser. Moderne immunsuppressiv behandling har redusert mortaliteten til under 10 % (4), men gir hyppige og alvorlige bivirkninger.
Paraneoplastisk pemfigus
Paraneoplastisk pemfigus er en sjelden tilstand som forekommer hos pasienter med malign grunnlidelse, som oftest av lymfoid type. Hudforandringene kan ha et mer varierende utseende enn ved pemphigus vulgaris. Munnslimhinnen er oftest affisert, og andre slimhinner kan også være det. Histologiske prøver og funn ved immunfluorescensundersøkelse er mer variable enn ved pemphigus vulgaris. Tilstanden er behandlingsresistent, og prognosen er dårlig og dels avhengig av grunnsykdommen (7).
Bulløst pemfigoid
Bulløst pemfigoid debuterer gjerne på huden med sterkt kløende, urticarialiknende eksantem og etter hvert danning av store, spente blemmer på normal eller erytematøs hud, ofte på fleksorsteder (Figur 1). Tilstanden er vanligere enn pemphigus vulgaris og forekommer hyppigst hos eldre. Hos opptil en tredel av pasientene er det også slimhinneaffeksjon oralt (1, 5) eller anogenitalt. Selv om blemmene i munnslimhinnen sitter dypere enn ved pemphigus vulgaris, sprekker de lett og etterlater seg sviende erosjoner (Figur 2). Tilheling skjer uten arrdanning. Patogenesen omfatter danning av autoantistoff mot proteiner i basalmembranen, noe som medfører subepitelial blemmedanning. Histologiske prøver og immunfluorescensundersøkelse viser typiske funn (Tabell 1). Bulløst pemfigoid preges av betydelig morbiditet, men prognosen er god. Tilstanden kan oftest kontrolleres ved hjelp av lokale eller systemiske steroider.
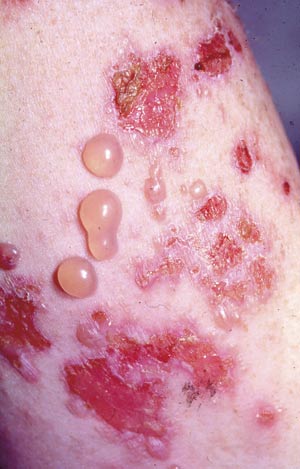
Figur 1. Bulløst pemfigoid i hud. Foto: Haukeland Universitetssjukehus.
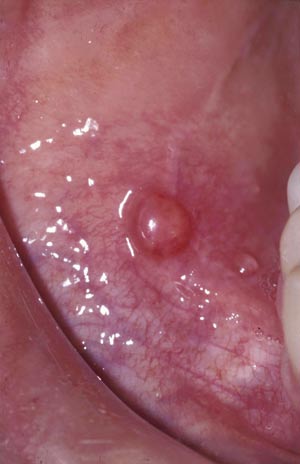
Figur 2. Bulløst pemfigoid med intakt blemme oralt. Foto: Bjarte Grung.
Slimhinnepemfigoid
Slimhinnepemfigoid (sikatrisielt pemfigoid) er en sjelden tilstand som alltid involverer munnslimhinnen og conjunctiva, men andre slimhinner affiseres også i varierende grad. Blemmene i munnslimhinnen er gjerne større enn ved bulløst pemfigoid. De etterlater seg smertefulle erosjoner som typisk tilheler med arrdanning. Sammenvoksninger mellom øyelokk og øyeeple (symblepharon) er en fryktet komplikasjon som kan føre til blindhet. Hudaffeksjon forekommer hos 30 – 50 % av pasientene, oftest i hode-hals-regionen og øverst på truncus (5, 6). Pasientene er typisk eldre kvinner.
Også ved denne tilstanden dannes det autoantistoffer mot proteiner i basalmembranen, men ikke de samme som ved bulløst pemfigoid. Histologiske prøver og immunfluorescensundersøkelse viser like funn ved begge tilstander (Tabell 1).
Slimhinnepemfigoid er mer behandlingsresistent enn bulløst pemfigoid. Risikoen for alvorlige komplikasjoner rettferdiggjør en mer aggressiv immunsuppressiv terapi (8).
Epidermolysis bullosa acquisita
Dette er en sjelden autoimmun sykdom. Det dannes antistoffer mot kollagen VII i basalmembransonen, og histologisk finnes subepitelial blemmedanning. Det kliniske bildet minner om det man ser ved hereditær epidermolysis bullosa. Tilstanden er ofte resistent mot immunsuppressiv behandling (9).
Dermatitis herpetiformis
Dermatitis herpetiformis er en ikke uvanlig autoimmun sykdom som i de aller fleste tilfeller er forbundet med glutensensitiv enteropati (cøliaki). Sykdommen forekommer hos voksne i alle aldersgrupper. Patogenesen er ukjent (10). Hudforandringene domineres typisk av regionalt utbredte, grupperte, ofte intenst kløende vesikler og blemmer. Hos 0 – 70 % av pasientene utvikles det etter hvert erosjoner i munnslimhinnen (5). Histologiske prøver og immunfluorescensundersøkelse viser typiske funn (Tabell 1). Dapson og glutenfri diett er viktigste behandlingstiltak. Prognosen er god.
Lineær IgA-dermatose
Dette er en sjelden autoimmun sykdom som forekommer både hos små barn (sykdommen ble tidligere betegnet som «chronic bullous disease of childhood») og hos voksne. Det kliniske bildet kan minne om dermatitis herpetiformis, men tilstanden er ikke assosiert med glutenintoleranse. Slimhinneforandringer finnes hos opptil 50 % av pasientene og varierer fra overflatiske erosjoner til mer omfattende orale forandringer. Konjunktival affeksjon forekommer (6). Histologiske prøver viser subepitelial blemmedanning som ved dermatitis herpetiformis, mens funn ved immunfluorescensundersøkelse er typisk for tilstanden (Tabell 1). Dapson og systemiske steroider er aktuelle behandlingstiltak. Prognosen er oftest god.
Andre blemmedannende tilstander
Erythema multiforme
Erythema multiforme er en nokså vanlig forekommende, akutt inflammatorisk sykdom som affiserer hud (erythema multiforme minor, 80 %), eventuelt hud og slimhinner (erythema multiforme major, 20 %). Tilstanden er i de fleste tilfeller trigget av infeksjon, hyppigst med herpes simplex-virus. Hos noen pasienter forekommer residiverende utbrudd.
Patogenesen er ikke endelig avklart, og både immunkompleksmekaniser og forsinket hypersensitivitet diskuteres. Det histologiske bildet er typisk. Hudaffeksjonen ved minorvarianten domineres av klassiske kokardelesjoner med symmetrisk utbredelse på distale deler av ekstremitetene. Initialt ses en erytematøs papel, og etter hvert utvikles kokarden med et mørkt sentrum omgitt av en blekere sone og utenfor denne en rødlig sone. Blemmedanning forekommer i alvorligere tilfeller. Ved erythema multiforme major er pasienten allment påvirket, og i tillegg til hudaffeksjonen foreligger orale, konjunktivale og ofte genitale lesjoner. I munnslimhinnen karakteriseres sykdommen av sårdanning, varierende fra solitære aftøse lesjoner til multiple overflatiske ulcerasjoner. Alle områder av munnslimhinnen kan være affisert.
Prognosen ved erythema multiforme er god, og forandringene går som oftest tilbake i løpet av 2 – 4 uker. Alvorlige tilfeller krever innleggelse i sykehus for systemisk steroidbehandling.
Stevens-Johnsons syndrom og toksisk epidermal nekrolyse
Disse betegnelsene har tradisjonelt vært benyttet om tilstander som er oppfattet som særlig alvorlige varianter av erythema multiforme. Nyere litteratur anfører derimot at erythema multiforme er en egen entitet som er patogenetisk, histologisk, klinisk og prognostisk forskjellig fra Stevens-Johnsons syndrom og toksisk epidermal nekrolyse (11). Hos barn er stafylokokkindusert subkorneal hudløsning (SSSS) en viktig differensialdiagnose.
Stevens-Johnsons syndrom og toksisk epidermal nekrolyse er typisk utløst av medikamenter som antiepileptika, antiflogistika, allopurinol og enkelte antibiotika. Det er alltid affeksjon av både hud og slimhinner. Klassiske kokardelesjoner forekommer ikke. I huden ses akutt nekrose ledsaget av blemmedanning og epidermolyse. Ved førstnevnte omfatter epidermolysen mindre enn 10 % av kroppsoverflaten, ved toksisk epidermal nekrolyse mer enn 30 %. Erosive og hemoragiske lesjoner samt ødem preger slimhinnene i munn, øyne, øvre luftveier, oesophagus og anogenitalt. Øyeaffeksjonen kan føre til blindhet. Pasienter med alvorlig Stevens-Johnsons syndrom og toksisk epidermal nekrolyse krever intensiv medisinsk behandling, gjerne i brannskadeavdeling.
Andre inflammatoriske tilstander
Lichen planus
Lichen planus er en inflammatorisk hud- og slimmhinnesykdom. Tilstanden er hyppig, med en prevalens på 0,3 – 2 % (12, 13). Sykdommen rammer middelaldrende, kvinner hyppigere enn menn. Kutan lichen planus er den vanligste kliniske formen, mens den orale varianten står for 15 – 35 % av tilfellene. Slimhinneaffeksjon forekommer hos opptil 65 % av pasientene med kutan lichen planus, og motsvarende finnes hudforandringer hos 15 – 30 % av dem med oral sykdom (14).
De klassiske hudforandringene er kantete rødligblå papler med glinsende overflate, ispedd tynne hvite linjer (Wickhams striae). Lesjonene sitter tett der huden har vært skadet (Köbners fenomen). Predileksjonsstedene er underarmenes volarsider og leggene (Figur 3). Sterk kløe krever oftest behandling med lokale eller systemiske steroider, men prognosen er uansett god med spontan tilbakegang av symptomene hos de fleste i løpet av 1 – 2 år.
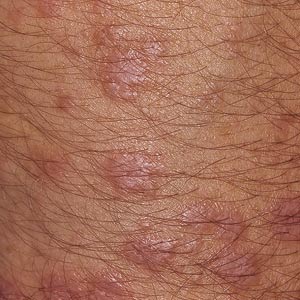
Figur 3. Lichen planus i hud. Foto: Bjarte Grung.
Oralt manifesterer tilstanden seg med flere ulike kliniske former, hvorav de viktigste er retikulær og erosiv lichen planus. Blandingsbilder forekommer hyppig. Den retikulære formen er vanligst og karakteriseres av et nettverk av hvite streker, oftest symmetrisk i kinnslimhinnene, tilsvarende Wickhams striae på hud (Figur 4). Tungeslimhinne, gingiva og lepper kan også være affisert. Ved erosiv oral lichen planus finner man sårdanning og betydelig inflammasjon og dermed sterkere symptomer (svie, smerter, vansker med fødeinntak) enn ved den retikulære formen. Øsofageal affeksjon forekommer. Oral lichen planus er ofte behandlingsresistent, men på sikt er spontan remisjon hyppig. Verdens helseorganisasjon har karakterisert tilstanden som prekankrøs, selv om det hersker uenighet om dette blant forskere (14, 15). Genitale slimhinnelesjoner forekommer hyppig (ca. 25 %) ved både kutan og oral lichen planus.
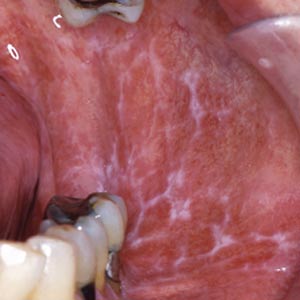
Figur 4. Retikulær oral lichen planus. Foto: Bjarte Grung.
Typiske funn ved histologiske prøver og immunfluorescensundersøkelse er viktig for å skille lichen planus fra andre lichenoide tilstander i hud og slimhinner, deriblant lichen sclerosus et atrophicus (tab 1). Genitale slimhinnelesjoner er kardinalfunnet ved sistnevnte. Hudlesjoner finnes hos 15 – 20 % av pasientene, mens oral affeksjon er uvanlig (16). Lichenoide lesjoner i munnslimhinnen kan også ses ved irritativ og kontaktallergisk stomatitt og ved transplantat-mot-vert-sykdom (graft versus host disease; GVHD). Disse kan i enkelte tilfeller ha et histologisk bilde som er sammenfallende med oral lichen planus. Kliniske funn vil da være avgjørende for diagnosen.
Psoriasis
Psoriasis er en vanlig forekommende tilstand, med prevalens på 1,5 – 2,5 % i vår del av verden. De fleste regner psoriasis som en isolert hudsykdom, med eller uten leddsymptomer, men flere forfattere omtaler orale lesjoner som en del av sykdomskomplekset (5, 17). En rekke studier viser overhyppighet av geografisk og fissurert tunge, ikke minst ved pustuløs psoriasis. Iblant kan man også finne histologisk påviste psoriatiske lesjoner i andre deler av munnslimhinnen.
Andre sykdommer som affiserer hud og munnslimhinne
Behçets sykdom
Behçets sykdom er en inflammatorisk multiorgansykdom som kjennetegnes av residiverende orale og genitale ulcerasjoner, uveitt og hudsymptomer i form av erythema nodosum, pseudofollikulitt og akneiforme lesjoner. Positiv kutan patergitest, dvs. pusteldanning etter nålestikk i huden, er hyppig, noe som kan brukes diagnostisk. Andre organer affiseres i varierende grad. Etiologien er ukjent. Sykdommen debuterer typisk i ung voksen alder, og begge kjønn rammes i lik grad. Tilstanden er ikke uvanlig i deler av Midtøsten, der det angis prevalenstall på 80 – 370 per 100 000 innbyggere (18), men er sjelden hos oss.
Munnhulelesjonene består av aftøse ulcerasjoner som ikke kan skilles fra dem ved «vanlig» aftøs stomatitt. Hudbiopsi kan vise vaskulitt i små kar, men er gjerne lite spesifikk. Man har liten nytte av immunfluorescensundersøkelse og måling av kjerneantistoffer i blod. Behandling med lokale steroider er ofte tilstrekkelig i lette tilfeller, mens multiorganaffeksjon krever systemisk immunsuppresjon. Prognosen er god i de fleste tilfeller, men yngre menn kan noen ganger utvikle alvorlig sentralnervøs vaskulitt som medfører betydelig morbiditet og mortalitet (18).
Lupus erythematosus
Lupus erythematosus er en relativt hyppig forekommende autoimmun sykdom som kan være primært systemisk eller lokalisert til hud som diskoid lupus erythematosus. Mellomformer som subakutt kutan lupus erythematosus forekommer også. Histologiske prøver og immunfluorescensundersøkelse (Tabell 1) viser typiske forandringer, og ved systemisk lupus erythematosus står påvisning av kjerneantistoffene ANA og anti-DNA sentralt i diagnostikken. Den systemiske varianten forekommer typisk hos yngre kvinner, mens forekomsten av diskoid lupus viser større variasjon når det gjelder kjønn og alder. Økt følsomhet for lys er vanlig ved lupus erythematosus, og hudforandringene forekommer gjerne på lyseksponerte områder, typisk som «sommerfuglutslett» i ansiktet. Mens utbredt, hissig og lett skjellende erytem er vanlig ved systemisk sykdom, preges den diskoide varianten av infiltrerte plakk med fastsittende skjelling og sentral atrofi.
Orale manifestasjoner forekommer hos opptil 80 % av pasientene med systemisk lupus (19), sjeldnere ved kutan sykdom. Lesjonene i munnslimhinnen består av ulcerasjoner eller erytematøse plakk med hvitlige striae som radierer fra periferien. Kinnslimhinne, gingiva og leppe er oftest affisert. Pasienter med systemisk lupus erythematosus krever særlig oppmerksomhet ved behandling hos tannlege (19). Behandlingen omfatter lokale og systemiske steroider, antimalariapreparater og systemiske immunsuppressive midler. Prognosen er utmerket ved diskoid lupus, variabel ved systemisk sykdom.
Autoimmune sykdommer som sklerodermi og dermatomyositt kan også gi symptomer fra både hud og munnslimhinne, men omtales ikke nærmere.
Diskusjon
Med unntak av lichen planus, erythema multiforme og lupus erythematosus er de beskrevne tilstandene sjeldent forekommende. Mens hudforandringene ofte er typiske, spiller de orale lesjonene på et mindre register, noe som kan gi diagnostiske problemer i de tilfeller hvor munnhuleforandringene forutgår de kutane. Det er også viktig å være oppmerksom på at blemmer i munnslimhinnen brister lett og kan etterlate erosive sårflater som eneste funn. Biopsier til vanlig histologisk undersøkelse og immunfluorescens (Tabell 1) spiller da en viktig rolle.
Hudbiopsi er en teknikk som beherskes av de fleste leger. Biopsering med hudstanse 4 mm eller større gir oftest et godt resultat. Biopsi fra munnslimhinne utføres oftest av spesialister i kjevekirurgi og munnhulesykdommer, oral medisin og kirurgi, periodonti eller øre-nese-halssykdommer. Biopsier til immunfluorescensundersøkelse må sendes som ferskt vev og etter spesielle rutiner.
Behandlingen ved sykdommer i munnslimhinnen er oftere enn ved hudsykdommer rent symptomatisk. På det norske markedet finnes kun ett lokalt steroidpreparat som er ment for bruk ved orale lesjoner (Kenacort-T munnsalve). Alvorlig inflammatorisk sykdom som affiserer munnslimhinnen indiserer ofte bruk av systemiske immunmodulerende eller immunsuppressive medikamenter.
Ettersom diagnostikk og behandling av tilstander som affiserer hud og munnslimhinne, ofte er krevende, og siden mange av sykdommene er sjeldne, er det ønskelig at spesielt interesserte og kompetente spesialistmiljøer tar hånd om slike pasienter. I tillegg til at nær kontakt mellom kliniker og patolog er essensielt, er dette et område som ligger vel til rette for et samarbeid mellom medisinske og odontologiske spesialiteter.
Hovedbudskap | |
|---|---|
• |
Mange sykdommer kan affisere både hud og munnslimhinne, men den kliniske presentasjonen er ofte ulik |
• |
Hudaffeksjonen kan komme forut for slimhinneforandringene og omvendt |
• |
Histologisk undersøkelse og immunfluorescensundersøkelse er viktige hjelpemidler i diagnostikken |
English summary
Holsen DS, Johannessen AC.
Diseases that affect both skin and oral mucosa.
338 – 43.
Background. Several diseases may affect both skin and oral mucosa. This article presents hereditary, autoimmune and other inflammatory conditions within this group.
Material and methods. Review of the literature as well as the authors’ own experience in the field.
Results and interpretation. Lichen planus, erythema multiforme and lupus erythematosus are relatively common disorders with generally favourable prognosis. The much less frequent bullous diseases pemphigoid and pemphigus are characterized by a high morbidity, often requiring a deliberate use of immunosuppressants. Stevens-Johnson syndrome, toxic epidermal necrolysis and the more severe forms of epidermolysis bullosa are potentially life-threatening conditions. It is recommendable that medical doctors and dentists are familiar with the clinical presentations and diagnostic procedures of diseases affecting skin and oral mucosa.
Litteratur
Regezi JA, Scuibba JJ, Jordan RCK. Oral pathology. Clinical pathologic correlations. 4. utg. St. Louis, MO: Saunders, 2003.
Champion RH, Burton JL, Burns DA et al, red. Rook/Wilkinson/Ebling Textbook of dermatology. London: Blackwell Science, 1998.
Has C, Kern JS, Bruckner-Tuderman L. Hereditäre Blasen bildende Hauterkrankungen. Hautarzt 2004; 55: 920 – 30.
Robinson JC, Lozada-Nur F, Frieden I. Oral pemphigus vulgaris. A review of the literature and a report on the management of 12 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1997; 84: 349 – 55.
Pindborg JJ. Diseases of the skin. I: Jones JH, Mason DK, red. Oral manifestations of systemic disease. 2. utg. London: Baillière Tindall, 1990.
Bickle KM, Roark TR, Hsu S. Autoimmune bullous dermatoses: a review. Am Fam Physician 2002; 65: 1861 – 70.
Allen CM, Camisa C. Paraneoplastic pemphigus: a review of the literature. Oral Diseases 2000; 6; 208 – 14.
Chan LS, Ahmed AR, Anhalt GJ et al. The first international consensus on mucous membrane pemphigoid. Arch Dermatol 2002; 138: 370 – 9.
Dabelsteen E. Molecular biological aspects of acquired bullous diseases. Crit Rev Oral Biol Med 1998; 9: 162 – 78.
Gjersvik PJ, Rønnevig JR. Dermatitis herpetiformis. Tidsskr Nor Lægeforen 2003; 123; 3234 – 6.
Bastuji-Garin S, Rzany B, Stern RS et al. Clinical classification of cases of toxic epidermal necrolysis, Stevens-Johnson syndrome, and erythema multiforme. Arch Dermatol 1993; 129: 92 – 6.
Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol 1991; 25: 593 – 619.
Dissemond J. Oral lichen planus: an overview. J Dermatol Treat 2004; 15: 136 – 40.
Eisen D. The clinical features, malignant potential, and systemic assosiations of oral lichen planus: a study of 723 patients. J Am Acad Dermatol 2002; 46: 207 – 14.
Pindborg JJ, Reichart PA, Smith CJ et al. Histological typing of cancer and precancer of the oral mucosa. World Health Organization Histological Classification of Tumours. Berlin: Springer-Verlag, 1997.
Powell JJ, Wojnarowska F. Lichen sclerosus. Lancet 1999; 353: 1777 – 83.
Zhu JF, Kaminski MJ, Pulitzer DR et al. Psoriasis: pathophysiology and oral manifestations. Oral Diseases 1996; 2: 135 – 44.
Al-Otaibi LM, Porter SR, Poate TWJ. Behçet’s disease: a review. J Dent Res 2005; 84: 209 – 22.
DeRossi SS, Glick M. Lupus erythematosus: considerations for dentistry. JADA 1998; 129: 330 – 9.
Adresse: Dag Sollesnes Holsen, Hudavdelingen, Haukeland Universitetssjukehus, 5021 Bergen. E-post: dag.holsen@helse-bergen.noArtikkelen har gjennomgått ekstern faglig vurdering.