Amelogenesis imperfecta: Gener, proteiner og fænotyper
uddannelsestandlæge, Kæbekirurgisk Afdeling, Odense Universitetshospital
tandlæge, Privat praksis
professor, overlæge, dr. med., Klinisk genetisk afdeling, Odense Universitetshospital, Syddansk Universitet
centerleder, specialtandlæge, Odontologisk Landsdels- og Videncenter, Aarhus Universitetshospital
tandlæge, odont.dr., pensionist, Umeå, Sverige
lektor, ph.d., Odontologisk Institut, Health, Tandlægeskolen i Aarhus,
Artikkelen har tidligere vært publisert i Tandlægebladet. 2013; 117: 574 - 82.
Introduktion - Traditionelt bliver amelogenesis imperfecta (AI) identificeret og klassificeret vha. emaljens kliniske udseende. Emaljen kan være misfarvet, blød og hypoplastisk af varierende grad samt udvise radiologisk forandring, ofte med nedsat røntgenkontrast. Nyere forskning muliggør inddeling af amelogenesis imperfecta på baggrund af genetiske fejl, hvilket bidrager til større forståelse af emaljeproteinernes funktion under amelogenesen samt sygdommens patogenese. Formål ? Denne artikel gennemgår gener og proteiner, hvor fejlkodning er kendt eller mistænkt for at deltage i AI-patogenesen. Proteinernes generelle funktioner bliver angivet kort i det omfang, de er kendte.
Materiale og metode - PubMed blev søgt uden sproglige restriktioner indtil 2012. Søgeord: amelogenesis imperfecta, amelogenesis, amelogenin, ameloblastin, amelin, sheathlin, enamelin, tuftelin, dentin sialophosphoprotein, dentin sialoprotein, dentin glycoprotein, dentin phosphoprotein, dentin matrix protein-1, enamelysin, kallikrein-4, dlx3 og kombinationer af disse.
Resultater - En række proteiner med vidt forskellig funktion ses involveret i amelogenesen. Den umodne emalje består af amelogeniner og non-amelogeniner. Hvis der er fejl i dannelse eller funktion af et eller flere af disse proteiner, vil det kliniske billede på sygdommen ofte afspejle, hvor i amelogenesens forløb fejlen opstår, og proteinets funktion betinger graden og karakteren af emaljemisdannelse.
Konklusion - Det tidspunkt af amelogenesen, der bliver forstyrret - altså den periode af emaljedannelsen, hvor en AI-gen- og proteinfejl fremtræder - afspejler sig direkte i den kliniske AI-fænotype med enten hypoplastisk, hypomineraliseret eller umoden emalje. Blandede AI-fænotyper kan forekomme.
Klinisk relevans
Det er en stor udfordring for tandlægen at diagnosticere amelogenesis imperfecta (AI), fordi AI er en sjælden tanddannelsesforstyrrelse, som optræder med stor fænotypisk variation. Derfor kan kendskab til den bagvedliggende genetik og patogenese være en hjælp i det diagnostiske arbejde. Samtidig repræsenterer AI en behandlingsmæssig udfordring for tandlægen, idet behandlingen, afhængigt af den kliniske manifestation, kan være alt fra fluorpenslinger og plast til fast protetik på samtlige tænder.
Amelogenesis imperfecta (AI) er en arveligt betinget sygdom, der afficerer begge tandsæt, og som er uden systemisk involvering. AI resulterer i emaljedefekter af forskellige kliniske genotyper (1). Da AI er arvelig, er tandsygdommens prævalens stærkt forøget i områder med ramte familier. Svenske studier har fundet en forekomst i Sydvestsverige på 0,3: 1000 (2), mens der i Nordsverige findes ca. 1,4: 1000 (3). I Danmark mangler vi evidens for forekomsten, men den skønnes at være i størrelsesordenen 0,125: 1000. AI inddeles klinisk i den hypoplastiske, den hypomineraliserede og den umodne type samt blandingstyper med taurodonti. Anglificerede synonymer anvendes også. «Hypocalcifikationstypen» er synonymt med «den hypomineraliserede type», og «hypomatureringstypen» er synonymt med «den umodne type». AI kan nedarves autosomalt recessivt, autosomalt dominant eller X-bundet (4 - 6). Formålet med denne artikel er at give en oversigt over gener og proteiner, der er involveret i amelogenese, samt deres funktion, for dermed at give et indblik i sygdommens patogenese.
Materiale og metode
PubMed blev søgt uden sproglige restriktioner frem til og med 2011. Søgeord: Amelogenesis imperfecta, amelogenesis, amelogenin, ameloblastin, amelin, sheathlin, enamelin, tuftelin, dentin sialophosphoprotein, dentin sialoprotein, dentin glycoprotein, dentin phosphoprotein, dentin matrix protein-1, enamelysin, kallikrein-4, dlx3 og kombinationer af disse.
Resultater
Amelogenese
Amelogenesen er en tretrins proces. Først aflejres emaljen i sin endelige tykkelse af ameloblasterne. Derefter passerer en stigende mængde mineraler ameloblastlaget og inkorporeres i emaljen. Endelig, inden emaljen færdigmineraliserer, bliver overskud af organisk materiale fjernet vha. enzymer udskilt fra ameloblasterne ( figur 1).
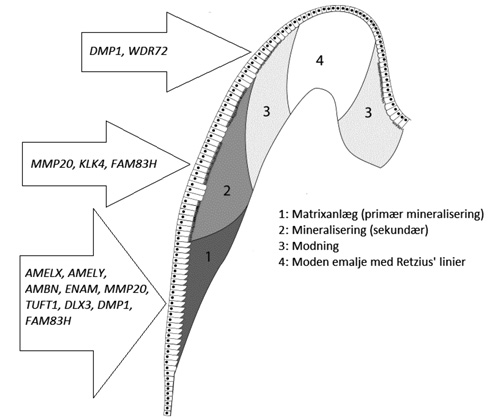
Figur 1. Skitse af amelogenese på den umodne tand, hvor emaljedannelsen er startet ved kusptoppen og her er i gang med den afsluttende mineralisering, mens der er sekretion af ny emaljematrix cervikalt. Illustrationen viser de enkelte emaljegeners udtryk under amelogenesens forskellige faser. Mutation i et emaljegen kan føre til, at proteinet bliver ødelagt. Det vil forstyrre den fase, hvori proteinet er aktivt. Emaljen bliver dannet i fire faser. 1) Sekretion af organisk matrix, der indeholder emaljeproteiner og er 30 % mineraliseret fra begyndelsen. 2) Mineralisering med øget tilførsel af mineral omkring emaljeproteinerne og mineralisering udefra. 3) Modning med øget fjernelse af organisk matrix og plads til mineralisering indefra. 4) Afsluttende hypermineralisering i emaljens overflade.
Selve aflejringen af emaljematrix er forudgået af en præsekretorisk fase, hvor ameloblasterne uddifferentieres fra det indre emaljeepitel til deres sekretoriske fænotype med en Tomes'' proces, ændret polaritet og et veludviklet proteinsynteseapparat. Under amelogenesen aflejrer ameloblasterne en emaljematrix med 70 % organisk materiale i et lag, der svarer til tykkelsen af den færdige emalje. Materialet i sekret-granula har en primær mineralisering med 30 % mineralindhold, som er oparbejdet, allerede inden de bliver udskilt fra ameloblasterne.
Emaljematrix aflejres uorganiseret i det inderste lag imod dentinen og i det yderste lag mod tandsækken, mens det mellemliggende lag er organiseret i emaljeprismer og interprismatisk emalje. Prismegrænserne fremstår som smalle zoner med et relativt højt indhold af organisk materiale.
Når aflejringen af emaljematrix er afsluttet, overgår ameloblasterne til mineraliserings- og modningsfasen og øger herved mineralindholdet i den allerede secernerede emaljematrix. Den afsluttende emaljedannelse sker igennem tre stadier; sekundær, tertiær og quarternær mineralisering ( figur 2 og tabel 1).
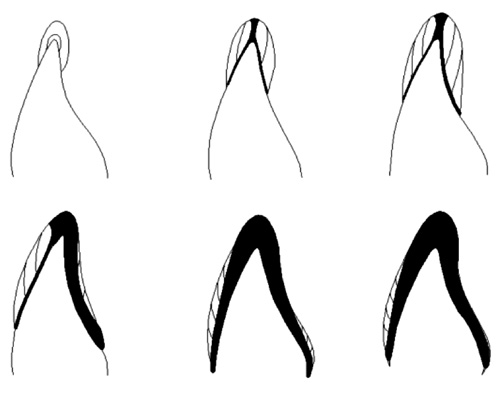
Figur 2. Den ufarvede emalje symboliserer sekretion (også kaldet primær mineralisering). Emaljematrix bliver først anlagt i sin fulde tykkelse over relativt kort tid. Den farvede emalje (sort) skitserer den efterfølgende mineralisering og til sidst afsluttende modning af emalje. Denne - viser et mere realistisk forløb end skitsen i Fig. 1. Den organiske del af emaljematrix bliver så nedbrudt af enzymer. Den afsluttende mineralisering begynder indefra emalje-dentin-grænsen og arbejder sig udad. Det er en proces, der strækker sig over længere tid end den initiale mineralisering.
Amelogenesefase |
Genfejl 1 |
AI-fænotype |
|---|---|---|
1. Sekretionsfasen |
® |
Hypoplastisk |
2. Mineraliseringsfase |
® |
Hypomineraliseret |
3. Modningsfase (fjernelse af protein/slutmineralisering) |
® |
Umoden |
4. Modningsfase (hypermineralisering af overflade) |
® |
Umoden |
I den sekundære mineraliseringsfase tilføres emaljen store mængder mineral. Mineralindholdet begynder først at stige tættest på ameloblastlaget, hvorefter mineralindholdet øges gradvist ind imod emalje-dentin-grænsen.
Den tertiære mineralisering, hvor stort set hele den organiske matrix fjernes og erstattes med mineral, begynder ved emalje-dentin-grænsen og arbejder sig ud mod ameloblastlaget. Derved øges mineralindholdet gradvist i emaljen til de endelige ca. 96 % (vægtprocent); resten udgøres af vand og protein.
I den kvarternære mineraliseringsfase foregår en hypermineralisering af det yderste lag af emalje, hvilket er den sidste ændring i emaljen, inden tanden bryder frem i mundhulen.
Efter mineraliseringens afslutning overgår ameloblasterne til en protektorisk fase, hvor de beklæder den nydannede emalje som det reducerede emaljeepitel. Når tanden bryder frem i mundhulen, fusionerer noget af det reducerede emaljeepitel med det gingivale epitel og danner kontaktepitelet, mens resten slides bort (10).
AI-typer og amelogenese
I begyndelsen af 1990''erne blev det påvist, at hvis der sker ændring af amelogenesen med mutation i et af de involverede gener (11), vil dette kunne medføre en klinisk ændring i emaljen. I dag kan vi se, at emaljeforandringen hos en AI-type i store træk er karakteristisk for den fase i amelogenesen, der er forstyrret (tabel 2).
AMELOGENESE | ||||
|---|---|---|---|---|
Faser |
1 |
2 |
3 |
4 |
Sekretion |
Mineralisering |
Modning |
Modning |
|
Ameloblaster |
Secernerer matrix-anlæg |
Transporterer mineral |
Fjerner protein og udskiller mineral |
|
Mineralisering |
Primær |
Sekundær |
Tertiær |
Quaternær |
Her secerneres emaljeproteiner, som fra starten er ca. 30 % mineraliseret |
Her aflejres flere mineraler i matrix, og fjernelse af protein bliver påbegyndt |
Emaljen mineraliserer videre, mens emaljeprotein i matrix bliver fjernet |
Afsluttende (hyper)mine-ralisering af emaljens overflade |
|
Emaljeanlægget er meget blødt |
Emaljen bliver hvid, blød og kridtet |
Emaljen bliver hård, transparent, mindre hvidlig |
Emaljen får normal hårdhed |
|
Genekspression* |
Amelogenin (AMELX, AMELY), Ameloblastin (AMBN), Enamelin (ENAM), Tuftelin (TUFT1), Enamelysin (MMP20), DLX3 (DLX3), Dentin sialoprotein (DSPP), Dentin phosphoprotein (DSPP), Dentin matrix protein-1 (DMP1) (FAM83H) |
Enamelysin(MMP20) Kallikrein-4(KLK4) (FAM83H) |
Dentin matrix protein-1 (DMP1), Kallikrein-4 (KLK4) (WDR72) |
|
AMELOGENESIS IMPERFECTA | ||||
Typer/Undergrupper |
(I) Hypoplastiske type: glat/ru hård generelt/lokale pits agenesi af emalje |
(II) Hypomineraliserede type: meget blød opaque |
(III) Umodne type: Evt. lidt blødopaque snow cap/pigment |
|
(IV) Blandingstyper: Taurodonti, mv. | ||||
Den hypoplastiske type
Hvis aflejringen af emaljematrix bliver forstyrret, bliver emaljen ufuldstændigt anlagt, og dermed hypoplastisk. Emaljen kan fx blive tynd og glat eller ru og ujævn ( - 3). Der kan ses folder, riller eller små pits, som ofte er misfarvede. Den hypoplastiske AI-type har manifestation i begge tandsæt, men optræder meget varieret. Tilstanden er relativt sjælden. Mineralindholdet i den anlagte emalje er overvejende normalt. Emaljen får derved normal hårdhed, og overfladen føles solid ved sondering. Der kan være radiologisk forandring, men oftest med normal kontrast mellem emalje og dentin.
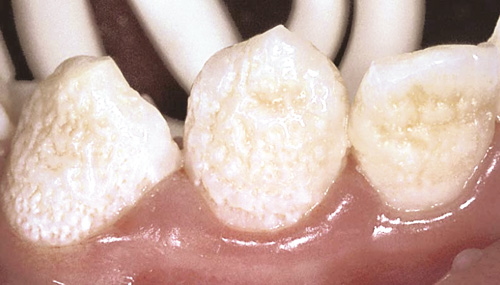
Figur 3. En ikke-fuldstændig sekretion af emaljematrix gør, at den hypoplastiske emalje er ru, ujævn og tyndere. Emaljen har her normal hårdhed og kan modstå både abrasion og sondering.
Den hypomineraliserede type
Ved denne type af AI er emaljematrix blevet fuldstændigt anlagt, men den sekundære mineralisering mangler, hvilket resulterer i lav røntgenkontrast mellem emalje og dentin. Emaljen er meget blød og går let tabt pga. almindelig tygning og attrition. Ved eruption er emaljen ofte gul-brun pga. interkrystallinsk misfarvning, og den kan evt. blive grålig til mørkebrun ( figur 4).
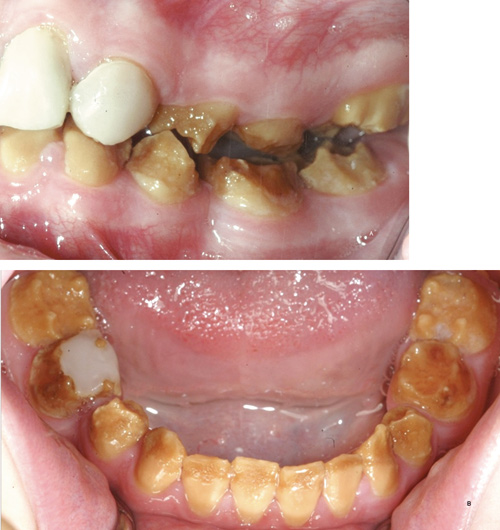
Figur 4. Her er emaljematrix anlagt i fuld tykkelse ved sekretion af ameloblasterne, men uden den efterfølgende sekundære mineralisering, og emaljen er hypomineraliseret og derfor meget blød og let at nedbryde.
Den umodne type
Emaljematrix i den umodne type er fuldstændigt anlagt og mineraliseret i højere grad, men dog uden fuldt afsluttet mineralisering. Emaljens hårdhed er kun lidt reduceret. Emaljeflager kan evt. springe af under funktion. Umoden emalje er et resultat af en forstyrrelse i modningsfasen, dvs. i forbindelse med enten den tertiære eller den kvarternære mineralisering. Hvis emaljedannelsen bliver forstyrret med små sprækker mellem emaljeprismerne, kan fødeemner evt. trænge ind i overfladen og give anledning til misfarvninger (12).
Proteiner involveret i amelogenesen
I forbindelse med dannelsen af tandens emalje udskilles der fra ameloblasterne en række proteiner, der alle spiller en særlig rolle for amelogenesen (tabel 2). Mutation i gener, der koder aminosyresekvensen i disse proteiner, kan bevirke, at emaljeproteinernes funktion ændres eller går tabt, hvilket er baggrund for optræden af AI. Emaljens proteiner består af ca. 90 % amelogenin og ca. 10 % såkaldte non-amelogeniner (10,13).
Amelogenin (AMELX og AMELY)
Amelogeninerne er en heterogen gruppe af store bipolære proteiner, der omgiver emaljekrystallerne og faciliterer deres længdevækst (14). Under den senere modning af emaljen fjernes amelogeninerne ved proteolytisk nedbrydning (7). De mange forskellige isoformer af amelogenin opstår pga. forskellige kløvninger af mRNA, der muliggøres af genets opbygning med syv exons eller ved enzymatisk modifikation af proteinet efter sekretion (15).
Amelogenin kodes af to forskellige gener på hhv. X-kromosomet (AMELX) og Y-kromosomet (AMELY). Amelogeningenerne AMELX og AMELY er lokaliseret til to steder hhv. kromosomregion Xp22.3-p22.1 (Online Mendelian Inheritance in Man (OMIM) #300391) og Yp11 (OMIM #410000).
Det betyder, at der er en mulig forskel i amelogeninmængde og -type mellem kønnene.
Én undersøgelse har vist, at 10 % af amelogenin hos mænd kommer fra Y-kromosomet (16).
Opstår der fejl i AMELX giver det anledning til den X-bundne recessive form af AI (OMIM #301200) (17).
Ved denne form er symptomerne mest udtalte hos mænd, som jo kun har ét X-kromosom. Mænd med X-bundet AI har en reduktion i funktionen af amelogenin på 90 %. Kvinder vil have en mosaik med gennemsnitligt 50 % tab af funktion (16,18). Denne mosaik er et interessant eksempel på den tilfældige inaktivering af det ene af kvindens X-kromosomer tidligt i fosterudviklingen, også kaldet lyoniserings-effekten. Det er derfor tilfældigt, om en ameloblast har det normale eller det muterede X-kromosom aktivt. Hos patienter med AI kan der som følge af denne X-kromosom-inaktivering opstå emaljefurer løbende langs tandens længdeakse, som svarer til ameloblaster med mutation i AMELX (19,20). Der er ikke beskrevet mutationer i AMELY som årsag til AI.
De kliniske fænotyper, der ses som følge af AMELX mutationer, inkluderer både de hypoplastiske, de hypomineraliserede, de umodne og de blandede typer (19 - 24).
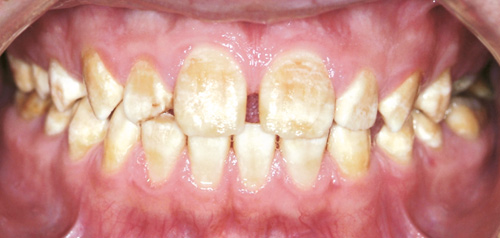
Figur 5. Her er emaljematrix anlagt i fuld tykkelse, og med efterfølgende sekundær mineralisering, men uden afsluttende modning af emaljen. Emaljen er her umoden og relativt hård, men emaljen kan ikke modstå, at farvestof fra føden trænger ind imellem emaljeprismerne og misfarver emaljen.
Non-amelogeninerne
Non-amelogeninerne er en heterogen gruppe indeholdende ameloblastin, enamelin, tuftelin, dentin sialoprotein, dentin glycoprotein, dentin phosphoprotein og dentin matrix protein-1, matrix metalloproteinase20 og kallikrein-4 (10,25).
Ameloblastin, amelin eller sheathlin (AMBN)
Ameloblastin udgør ca. 5 % af det totale protein i emaljematrix (26). Ameloblastin nedbrydes hurtigt efter sekretionen i flere dele, hvoraf den ene gruppe hurtigt nedbrydes helt. Ameloblastins normale fysiologiske rolle i forbindelse med amelogenesen er indtil videre ukendt (27). Dyreeksperimentielle studier, der anvender transgene mus, har vist, at en overproduktion af ameloblastin giver en emalje, der domineres af interprismatisk emalje og mangler det yderste prismeløse emaljelag (28). Genet for ameloblastin (AMBN) er lokaliseret til 4q21, tæt på genet for enamelin (ENAM), og er kandidatgen for en autosomal form af AI. Der er dog indtil videre ikke beskrevet mutationer i AMBN som årsag til AI. I en transgen musemodel, hvor genet for ameloblastin overudtrykkes, ses en AI-lignende fænotype (28).
Enamelin (ENAM)
Enamelin udgør ca. 2 % af det totale proteinindhold i emaljematrix (26). Proteinet binder sig til mineral i emaljen og er forbundet med emaljekrystallers kimdannelse (nukleation) og vækst. Enamelins funktion i forbindelse med amelogenesen er tilsyneladende relateret til den forlængelse af emaljekrystaller, der finder sted under emaljens vækst i tykkelse (29).
ENAM er lokaliseret til 4q21, og mutation i dette gen kan give anledning til både autosomalt dominant og autosomalt recessiv AI. I begge tilfælde kan der være tale om AI af den hypoplastiske, den hypomineraliserede eller den umodne type (29 - 31). Den hypoplastiske type kan optræde både generelt og lokaliseret (32).
Ved den autosomale recessive form af AI, som opstår som følge af en mutation i ENAM, vil personer, der er heterozygote og dermed anlægsbærere, have en mild version af AI med focale emaljehypoplasier og/eller misfarvninger. Er patienten derimod homozygot for mutation i ENAM, eller er der tale om en mutation, der medfører dominant AI, er den resulterende fænotype en mere alvorlig form for AI. Denne er karakteriseret ved, at patientens tænder udviser generel hypoplasi af emaljen, og at den tilstedeværende emalje er hypomineraliseret. Desuden er AI hos den homozygote patient ofte associeret med en malokklusion med åbent bid (30,31).
Tuftelin (TUFT1)
Tuftelin findes i små mængder omkring emalje-dentin-grænsen og menes at have betydning for dannelsen af denne, men proteinet er ikke specifikt lokaliseret til emaljen og findes i mange vævstyper (10). Genet, som koder for tuftelin, kaldes TUFT1 (er lokaliseret til 1q21 - 1q31) (33). Mutationer i TUFT1 er endnu ikke observeret som årsag til AI.
Dentin sialophosphoprotein (DSPP)
Genet for dentin sialophosphoprotein DSPP er lokaliseret til 4q21.3 (OMIM #125485) (34,35). Proteinet dannes fra sekretoriske odontoblaster og præsekretoriske ameloblaster (36 - 38). Dentin sialophosphoprotein har betydning for normal dentinogenese, og mutationer i genet kan medføre dentinogenesis imperfecta (39,40). Betydningen for normal amelogenese er endnu uklar. Proteinet bliver postsekretorisk spaltet i tre dele: Dentin sialoprotein, dentin glycoprotein og dentin phosphoprotein (5,21,34).
Dentin sialoprotein (DSP)
Ved overekspression af dentin sialoprotein opstår der et aprismatisk emaljelag ved emalje-dentin-grænsen med forøget tykkelse, hvorfor dentin sialoprotein sandsynligvis spiller en rolle ved etableringen af emalje-dentin-grænsen (41).
Dentin glycoprotein (DGP)
Funktion og eventuelt betydning for amelogenesen er endnu ukendt (25).
Dentin phosphoprotein (DPP)
Funktion og eventuelt betydning for amelogenesen er endnu ukendt, men ved overekspression af dentin phosphoprotein ses en fænotype med en hypoplastisk, kridtet og prismeløs emalje, der let slides bort (41).
Dentin matrix protein-1 (DMP1)
DMP1 er også lokaliseret til 4q21 (OMIM #600980), et område, der er associeret med forstyrrelser i emaljedannelsen. DMP1 er således kandidatgen for AI. Flere gener, der koder for emaljeproteiner, er lokaliseret her. Proteinets funktion er indtil videre ukendt, men det kædes sammen med mineralisering, idet studier har vist, at Dmp1 udtrykkes kortvarigt og svagt i sekretoriske ameloblaster og kraftigt i modningsameloblaster samt i odontoblaster og osteoblaster (42).
Mutation i DMP1 kan medføre recessiv hypofosfatæmisk rakitis, og ved koblingsbaserede studier er det fundet at være kandidatgen for dentinogenesis imperfecta. Der er endnu ingen rapporter om Dmp1-mutationer som kausativ faktor ved AI (43,44).
Enamelysin (MMP20)
Enamelysin er en Ca++ afhængig metalloproteinase, der udøver sin funktion i forbindelse med sekretionsfasen og den tidlige modningsfase i amelogenesen. MMP20 er lokaliseret til 11q22-q23 (OMIM # 604629). Enamelysin er et tandspecifikt enzym, der er ansvarligt for nedbrydningen af proteiner i forbindelse med amelogenesen, herunder amelogenin, det dominerende protein i amelogenesen (45). Enamelysin er desuden ansvarligt for at aktivere kallikrein-4 enzymet (46). Mutation i MMP20 kan give anledning til autosomal recessiv AI med hypomineraliseret og hypoplastisk emalje (45).
Kallikrein-4 (KLK4)
Kallikrein-4 er en Ca++ uafhængig serin protease, der aktiveres af enamelysin efter sekretionen (46). Kallikrein-4 udøver sin funktion i ameloblasternes hvilefase og igennem hele modningsfasen, hvor det nedbryder proteiner og deres rester. KLK4 nedbryder bl.a. amelogenin. Denne nedbrydning er en direkte forudsætning for, at amelogenin fjernes fra emaljematrix og dermed giver plads til optag af yderligere mineral i emaljen under emaljekrystallernes breddevækst (46). KLK4 er lokaliseret til 19q13.3-q13.4. Fejl i KLK4 kan medføre en autosomal recessiv form af AI af den hypomineraliserede type (46).
DLX3
Genet DLX3 er en del af D11-homeoboxgenerne, som spiller en væsentlig rolle for udviklingen af mange af kroppens væv, inklusive kraniet, tænder, hjerne, hår og nerver. DLX3 proteinet er en transskriptionsfaktor, der styrer ekspressionen af andre gener. DLX3 er lokaliseret til 17q21.3-q22.
Mutationer i DLX3 kan klinisk medføre en AI af blandingstype med både hypoplastisk og umoden emalje samt taurodontisme (47,48).
TYPE |
Fænotype |
Træk |
Arvegang |
|---|---|---|---|
IA |
Hypoplastisk |
Pits generelt |
Autosomal dominant |
IB |
Hypoplastisk |
Pits lokalt |
Autosomal dominant |
IC |
Hypoplastisk |
Pits lokalt |
Autosomal recessiv |
ID |
Hypoplastisk |
Opaque glat |
Autosomal dominant |
IE |
Hypoplastisk |
Opaque glat |
X-bundet dominant |
IF |
Hypoplastisk |
Opaque ru |
Autosomal dominant |
IG |
Hypoplastisk |
Emalje agenesi |
Autosomal recessiv |
IIA |
Umoden |
Opaque pigmenteret |
Autosomal recessiv |
IIB |
Umoden |
Opaque |
X-bundet recessiv |
IIC |
Umoden |
Snow caps |
X-bundet |
IID |
Umoden |
Snow caps |
Autosomal dominant? |
IIIA |
Hypomineraliseret |
Opaque |
Autosomal dominant |
IIIB |
Hypomineraliseret |
Opaque |
Autosomal recessiv |
IVA |
Umoden-hypoplastisk |
Med taurodonti |
Autosomal dominant |
IVB |
Hypoplastisk-umoden |
Med taurodonti |
Autosomal dominant |
Non-amelogeninerne med ukendt funktion
De senest rapporterede gener, som er involveret i amelogenesen, og hvori mutationer er associeret med AI, koder for proteiner, hvis funktion endnu er ukendt.
Fam83H
Genet Fam83H, beliggende 8q24.3 (OMIM #130190), koder for et 1.179 aminosyre stort protein, hvis rolle i amelogenesen er ukendt. Fam83H udtrykkes i såvel odontoblaster som ameloblaster, og mindst ni forskellige mutationer i dette gen er associeret med autosomal dominant AI af den hypomineraliserede type (49). Associationen mellem Fam83H og AI er første gang rapporteret i 2008 (50), og mutationer i dette gen har vist sig at forklare en meget stor andel af rapporterede AI-populationer, herunder også i to danske familier (51,52). Mutationerne medfører, at det dannede protein forkortes betydeligt. Fænotypisk kan ses generaliserede hypomineraliseringer med meget svag og abnorm emalje, mens der ved lidt mindre trunkering kan ses mere lokaliseret hypomineralisering overvejende i den cervikale del af tandkronen.
WDR72
Genet WDR72, der er placeret på kromosom 15, 15q21.3, er associeret med recessiv AI af umoden type. (OMIM #6132124). WDR72 udtrykkes specielt i modningsameloblaster. Emaljelaget har ved eruption normal tykkelse, er gulligt opaque, og under funktion optræder tab af emalje samt brunlig misfarvning (53).
Gener og proteiner styrer emaljens udvikling
Initieringen af emaljekrystaller ved emalje-dentin-grænsen er associeret med expression af DSPP og de strukturelle emaljeproteiner AMELX, AMELY, ENAM, AMBN og MMP20. Emaljeformationen fortsætter ved elongering af emaljekrystaller i mineraliseringsfronten lige under ameloblastens distale ende, hvor disse proteiner bliver secerneret. Når krystal-elongeringen er komplet, og emaljelaget når sin endelige tykkelse, vil KLK4 facilitere nedbrydning og reabsorption af akkumulerede emaljeproteiner. Betydningen af disse matrix-proteiner er bedst understreget af de dramatisk ændrede emalje-fænotyper, der ses i gen-modificerede mus med knockout af emaljematrix-gener (DSPP, AMELX, AMBN, MMP20) og i familier, hvor mutationer er identificeret i emaljematrixgenerne DSPP, AMELX, ENAM, MMP20 eller KLK4. På baggrund af andre tilsvarende dyreundersøgelser er det foreslået, at Fam83H spiller en rolle tidligt i amelogenesen under differentieringen af preameloblasten til den funktionsdygtige ameloblast. Mutationer i dette gen har derfor voldsom indflydelse på emaljens fænotype (54).
Konklusion
Det er i dag muligt at beskrive, hvordan forstyrrelser og sekvensvariationer i emaljegener kan medføre ekspression af ændrede emaljeproteiner. Flere forskellige genfejl kan føre til amelogenesis imperfecta. Afhængigt af hvilken fase i amelogenesen der bliver forstyrret - sekretion, mineralisering eller modning - vil dette medføre en karakteristisk forandring i emaljen, som overvejende er af den hypoplastiske, hypomineraliserede eller den umodne type; blandingstyper kan også forekomme.
Taksigelse
Tak til Pia Kjærgaard Karlsen for hjælp til figur 2.
English summary
Jakobsen C, Skou R, Hertz JM, Gjørup H, Bäckman B, Løvschall H.
Amelogenesis imperfecta: Genes, proteins, and phenotypes
Nor Tannlegeforen Tid 2013; 123: 712-19
Introduction - Traditionally, amelogenesis imperfecta (AI) is identified and classified based on changes in the clinical appearance of the enamel. The enamel may be discolored, soft and hypoplastic to varying degrees, and exhibit radiological changes often with reduced X-ray contrast. Recent research enables classification of amelogenesis imperfecta on the basis of genetic defects, which contributes to a better understanding of both the enamel protein function during amelogenesis, and the pathogenesis of the disease.
Objective - This article reviews genes and proteins, in which error coding is known or suspected to participate in the AI pathogenesis. The general protein functions are listed briefly to the extent that are known.
Materials and method - PubMed was searched without language restrictions until 2012. Keywords: amelogenesis imperfecta, amelogenesis, amelogenin, ameloblastin, amelin, sheathlin, enamelin, tuftelin, dentin sialophosphoprotein, dentin sialoprotein, dentin glycoprotein, dentin phosphoprotein, dentin matrix protein-1, enamelysin, kallikrein-4, dlx3 and combinations of these.
Results - A number of proteins with different function are involved in amelogenesis. The immature enamel contain amelogenins and non-amelogenins. If there are errors in protein formation or function, of one or more of these proteins, the clinical representation of the disease often reflects the stage during amelogenesis where the fault show up. Changes of the protein function determine the degree and nature of the enamel malformation.
Conclusion - The phase of enamel formation where an AI gene and protein error appears and amelogenesis is disturbed - during stages of respectively secretion, mineralization or maturation of enamel matrix - is mirrored directly in the clinical AI phenotypes with either hypoplastic, hypomineralisation, or hypomaturation enamel type. Mixed AI types may appear.
uddannelsestandlæge, Kæbekirurgisk Afdeling, Odense Universitetshospital
tandlæge, Privat praksis
professor, overlæge, dr. med., Klinisk genetisk afdeling, Odense Universitetshospital, Syddansk Universitet
centerleder, specialtandlæge, Odontologisk Landsdels- og Videncenter, Aarhus Universitetshospital
tandlæge, odont.dr., pensionist, Umeå, Sverige
lektor, ph.d., Odontologisk Institut, Health, Tandlægeskolen i Aarhus,
Artikkelen har tidligere vært publisert i Tandlægebladet. 2013; 117: 574 - 82.
Litteratur
Witkop CJ, Jr. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J Oral Pathol. 1988; 17: 547 - 53.
Sundell S, Koch G. Hereditary amelogenesis imperfecta. I. Epidemiology and clinical classification in a Swedish child population. Swed Dent J. 1985; 9: 157 - 69.
Bäckman B, Holm AK. Amelogenesis imperfecta: prevalence and incidence in a northern Swedish county. Community Dent Oral Epidemiol. 1986; 14: 43 - 7.
Witkop CJ Jr. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J Oral Pathol. 1988; 17: 547 - 53.
Aldred M, Crawford PJ, Savarirayan R et al. It''s only teeth - are there limits to genetic testing? Clin Genet. 2003; 63: 333 - 9.
Bäckman B. Inherited enamel defects. Ciba Found Symp. 1997; 205: 175 - 82.
Simmer JP, Hu JC. Dental enamel formation and its impact on clinical dentistry. J Dent Educ. 2001; 65: 896 - 905.
Pindborg JJ. Aetiology of developmental enamel defects not related to fluorosis. Int Dent J. 1982; 32: 123 - 34.
Larsen MJ, Richards A. Fluorids farmakologi, toksikologi og virkninger. Tandlægebladet. 2000; 104: 94 - 103.
Nancy A. Enamel: composition, formation and structure. In: Nancy A, ed. Ten Cate''s oral histology: development, structure and function. 6th ed. St. Louis: Mosby, 2003; 145 - 91.
Lagerström M, Dahl N, Nakahori Y et al. A deletion in the amelogenin gene (AMG) causes X-linked amelogenesis imperfecta (AIH1). Genomics. 1991; 10: 971 - 5.
Simmer JP, Hu JC. Dental enamel formation and its impact on clinical dentistry. J Dent Educ. 2001; 65: 896 - 905.
Lyaruu DM, Hu CC, Zhang C et al. Derived protein and cDNA sequences of hamster amelogenin. Eur J Oral Sci. 1998; 106 (Supp 1): S299 - 307.
Snead ML, Lau EC, Zeichner-David M et al. DNA sequence for cloned cDNA for murine amelogenin reveal the amino acid sequence for enamel-specific protein. Biochem Biophys Res Commun. 1985; 129: 812 - 8.
Simmer JP. Alternative splicing of amelogenins. Connect Tissue Res. 1995; 32: 131 - 6.
Salido EC, Yen PH, Koprivnikar K et al. The human enamel protein gene amelogenin is expressed from both the X and the Y chromosomes. Am J Hum Genet. 1992; 50: 303 - 16.
Collier PM, Sauk JJ, Rosenbloom SJ et al. An amelogenin gene defect associated with human X-linked amelogenesis imperfecta. Arch Oral Biol. 1997; 42: 235 - 42.
Crawford PJ, Aldred MJ. X-linked amelogenesis imperfecta. Presentation of two kindreds and a review of the literature. Oral Surg Oral Med Oral Pathol. 1992; 73: 449 - 55.
Lench NJ, Brook AH, Winter GB. SSCP detection of a nonsense mutation in exon 5 of the amelogenin gene (AMGX) causing X-linked amelogenesis imperfecta (AIH1). Hum Mol Genet. 1994; 3: 827 - 8.
Lench NJ, Winter GB. Characterisation of molecular defects in X-linked amelogenesis imperfecta (AIH1). Hum Mutat. 1995; 5: 251 - 9.
Kindelan SA, Brook AH, Gangemi L et al. Detection of a novel mutation in X-linked amelogenesis imperfecta. J Dent Res. 2000; 79: 1978 - 82.
Ravassipour DB, Hart PS, Hart TC et al. Unique enamel phenotype associated with amelogenin gene (AMELX) codon 41 point mutation. J Dent Res. 2000; 79: 1476 - 81.
Hart PS, Aldred MJ, Crawford PJ et al. Amelogenesis imperfecta phenotype-genotype correlations with two amelogenin gene mutations. Arch Oral Biol. 2002; 47: 261 - 5.
Kim JW, Simmer JP, Hu YY et al. Amelogenin p. M1T and p. W4S mutations underlying hypoplastic X-linked amelogenesis imperfecta. J Dent Res. 2004; 83: 378 - 83.
Yamakoshi Y, Hu JC, Fukae M et al. Dentin glycoprotein: the protein in the middle of the dentin sialophosphoprotein chimera. J Biol Chem. 2005; 280: 17472 - 9.
Stephanopoulos G, Garefalaki ME, Lyroudia K. Genes and related proteins involved in amelogenesis imperfecta. J Dent Res. 2005; 84: 1117 - 26.
Brookes SJ, Kirkham J, Shore RC et al. Amelin extracellular processing and aggregation during rat incisor amelogenesis. Arch Oral Biol. 2001; 46: 201 - 8.
Paine ML, Wang HJ, Luo W et al. A transgenic animal model resembling amelogenesis imperfecta related to ameloblastin overexpression. J Biol Chem. 2003; 278: 19447 - 52.
Hu JC, Yamakoshi Y. Enamelin and autosomal-dominant amelogenesis imperfecta. Crit Rev Oral Biol Med. 2003; 14: 387 - 98.
Hart TC, Hart PS, Gorry MC et al. Novel ENAM mutation responsible for autosomal recessive amelogenesis imperfecta and localised enamel defects. J Med Genet. 2003; 40: 900 - 6.
Kida M, Ariga T, Shirakawa T et al. Autosomal-dominant hypoplastic form of amelogenesis imperfecta caused by an enamelin gene mutation at the exon-intron boundary. J Dent Res. 2002; 81: 738 - 42.
Mårdh CK, Bäckman B, Holmgren G et al. A nonsense mutation in the enamelin gene causes local hypoplastic autosomal dominant amelogenesis imperfecta (AIH2). Hum Mol Genet. 2002; 11: 1069 - 74.
Deutsch D, Palmon A, Young MF et al. Mapping of the human tuftelin (TUFT1) gene to chromosome 1 by fluorescence in situ hybridization. Mamm Genome. 1994; 5: 461 - 2.
Yamakoshi Y, Hu JC, Fukae M et al. Dentin glycoprotein: the protein in the middle of the dentin sialophosphoprotein chimera. J Biol Chem. 2005; 280: 17472 - 9.
Feng JQ, Luan X, Wallace J et al. Genomic organization, chromosomal mapping, and promoter analysis of the mouse dentin sialophosphoprotein (Dspp) gene, which codes for both dentin sialoprotein and dentin phosphoprotein. J Biol Chem. 1998; 273: 9457 - 64.
Papagerakis P, Berdal A, Mesbah M et al. Investigation of osteocalcin, osteonectin, and dentin sialophosphoprotein in developing human teeth. Bone. 2002; 30: 377 - 85.
MacDougall M, Nydegger J, Gu TT. Developmental regulation of dentin sialophosphoprotein during ameloblast differentiation: a potential enamel matrix nucleator. Connect Tissue Res. 1998; 39: 25 - 37.
Bégue-Kirn C, Krebsbach PH, Bartlett JD et al. Dentin sialoprotein, dentin phosphoprotein, enamelysin and ameloblastin: tooth-specific molecules that are distinctively expressed during murine dental differentiation. Eur J Oral Sci. 1998; 106: 963 - 70.
Xiao S, Yu C, Chou X et al. Dentinogenesis imperfecta 1 with or without progressive hearing loss is associated with distinct mutations in DSPP. Nat Genet. 2001; 27: 201 - 4.
Zhang X, Zhao J, Li C et al. DSPP mutation in dentinogenesis imperfecta Shields type II. Nat Genet. 2001; 27: 151 - 2.
Paine ML, Luo W, Wang HJ et al. Dentin sialoprotein and dentin phosphoprotein overexpression during amelogenesis. J Biol Chem. 2005; 280: 31991 - 8.
D''Souza RN, Cavender A, Sunavala G et al. Gene expression patterns of murine dentin matrix protein 1 (Dmp1) and dentin sialophosphoprotein (DSPP) suggest distinct developmental functions in vivo. J Bone Miner Res. 1997; 12: 2040 - 9.
MacDougall M, Gu TT, Simmons D. Dentin matrix protein-1, a candidate gene for dentinogenesis imperfecta. Connect Tissue Res. 1996; 35: 267 - 72.
D''Souza RN, Cavender A, Sunavala G et al. Gene expression patterns of murine dentin matrix protein 1 (Dmp1) and dentin sialophosphoprotein (DSPP) suggest distinct developmental functions in vivo. J Bone Miner Res. 1997; 12: 2040 - 9.
Caterina JJ, Skobe Z, Shi J et al. Enamelysin (matrix metalloproteinase 20)-deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem. 2002; 277: 49598 - 604.
Hart PS, Hart TC, Michalec MD et al. Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet. 2004; 41: 545 - 9.
Dong J, Amor D, Aldred MJ et al. DLX3 mutation associated with autosomal dominant amelogenesis imperfecta with taurodontism. Am J Med Genet A. 2005; 133A: 138 - 41.
Zhao Z, Stock D, Buchanan A et al. Expression of Dlx genes during the development of the murine dentition. Dev Genes Evol. 2000; 210: 270 - 5.
Kim JW, Lee SK, Lee ZH et al. FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am J Hum Genet. 2008; 82: 489 - 94.
Mendoza G, Pemberton TJ, Lee K et al. A new locus for autosomal dominant amelogenesis imperfecta on chromosome 8q24. 3. Hum Genet. 2007; 120: 653 - 62.
Wright JT, Torain M, Long K et al. Amelogenesis imperfecta: genotype-phenotype studies in 71 families. Cells Tissues Organs. 2011; 194: 279 - 83.
Haubek D, Gjørup H, Jensen LG et al. Limited phenotypic variation of hypocalcified amelogenesis imperfecta in a Danish five-generation family with a novel FAM83H nonsense mutation. Int J Paediatr Dent. 2011; 21: 407 - 12.
El-Sayed W, Parry DA, Shore RC et al. Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am J Hum Genet. 2009; 85: 699 - 705.
Lee MJ, Lee SK, Lee KE et al. Expression patterns of the Fam83h gene during murine tooth development. Arch Oral Biol. 2009; 54: 846-50.
Adresse: Henrik Løvschall, Department of Dentistry, HEALTH, Aarhus University, Vennelyst Boulevard 9, 8000 Aarhus C, Danmark. E-post: loev@odont.au.dk
Artikkelen har gjennomgått ekstern faglig vurdering.
Jakobsen C, Skou R, Hertz JM, Gjørup H, Bäckman B, Løvschall H. Amelogenesis imperfecta: Gener, proteiner og fænotyper. Nor Tannlegeforen Tid. 2013; 123: 712-19