Neurobiology: General considerations - from acute to chronic pain
The understanding of the molecular mechanisms behind the transmission and modulation of pain, as well as of the brain circuitry involved in the processing of sensory discriminative, affective, cognitive and motivational components of pain, has increased dramatically during recent years. In the present article we give an outline of the pathways and brain regions involved in pain processing, the most important mediators, receptors and ion channels, as well as peripheral and central mechanisms for increased pain sensitivity and chronification of pain. We also touch on the striking similarities between the way we process physical and emotional pain, and the accumulating evidence that severe physical and social pain may alter brain function and that such changes may contribute to the transition from acute to chronic pain and to the comorbidity between chronic pain, depression and anxiety disorders. We also provide some practical tips to reduce pain, based on the recent neurobiological research, hand in hand with firm clinical experience.
Headlines | |
|---|---|
· |
Pathways and molecular mechanisms involved in the transmission and modulation of pain. |
· |
Processing of sensory discriminative, affective, cognitive and motivational components of pain. |
· |
Mechanisms for increased pain sensitivity and chronification of pain. |
· |
Similarities between physical and emotional pain. |
· |
Comorbidity between chronic pain, depression and anxiety disorders. |
Pain - a significant part of life - still an elusive figure
Every time we feel pain, it is an individual and highly subjective experience. Like love, hunger and anger, pain cannot be measured in quantitative (metric) terms. It can only, at best, be broadly assessed by ourselves or someone that can interpret our appearance and behavior in connection to pain. Both the experience of pain and the responses to pain are multidimensional involving sensory discriminative, affective, cognitive and motivational components. For example memory processes and behavioral changes are powerfully driven by pain experiences (1). It is therefore not surprising that information about pain is handled by a number of brain regions, which are also involved in our handling of many other aspects of our mental life. Furthermore, we are endowed with numerous mechanisms that may alter pain sensitivity, both in acute situations like most cases of toothache, and in chronic pathological pain syndromes like temporomandibular joint disorders (TMD). In the present article we try to describe some aspects of peripheral and central pain processing and pathological changes of possible relevance for the development and treatment, and in best case even the prevention, of chronic pain.
Pathways and brain regions involved in pain processing
Peripheral nociceptive fibers
The «pain sensors», the nociceptors, in for example the dental pulp and oral mucosa, are the free nerve endings of primary afferent fibers. There are two types of peripheral nociceptive («pain») fibers, A??fibers, that are myelinated with medium diameter, and C-fibers, that are thin and unmyelinated. A??fibers mediate the fast (first) pain, whereas C-fibers mediate the slow (second) and poorly localized pain, in response to an acute nociceptive stimulus. Like all other primary sensory neurons, the C- and A?-fibre-neurons have their cell bodies in the dorsal root ganglia (DRG) and the trigeminal ganglion (innervation of the mouth and face). The central endings of these neurons make synaptic contacts with neurons in the dorsal horn of the spinal cord and in the spinal trigeminal complex, respectively (Figure 1).
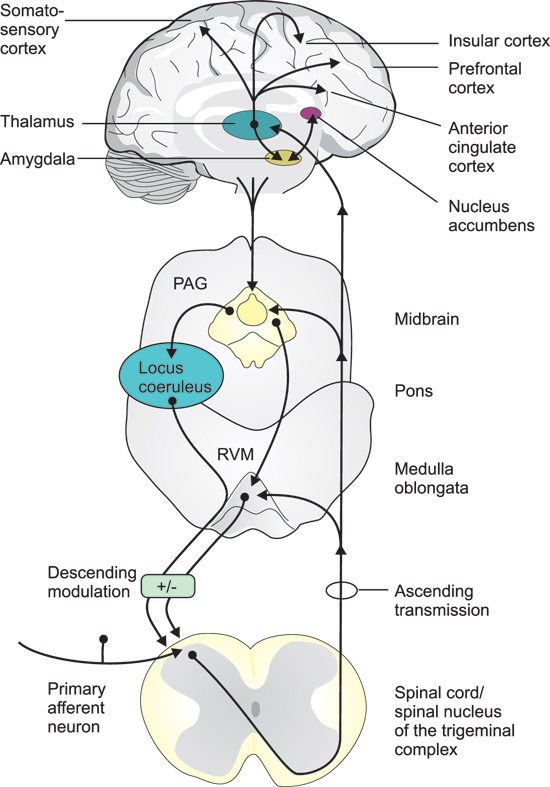
Figure 1. Pathways and brain regions involved in the transmission and modulation of pain signals. Primary afferent neurons, with their cell bodies in the dorsal root ganglia, or trigeminal ganglion (mouth and face), couple over to secondary neurons in the dorsal horn of the spinal cord, or the spinal nucleus of the trigeminal complex. The axons of the second order neurons cross the midline and project to the thalamus and a variety of other targets in the medulla oblongata, pons and mid brain, including the rostral ventral medulla (RVM) and the periaqueductal grey (PAG). Third order neurons, with their cell bodies in the thalamus, project to the somatosensory cortex, responsible for the sensory-discriminative aspects (intensity, location and quality) of pain, and to limbic cortical areas, such as the anterior cingulate, insular, and the prefrontal cortex involved in the mediation of the affective/emotional components (aversiveness) of pain. Thalamic neurons also project to the amygdala, which in turn interacts with the nucleus accumbens, a region involved in both pain processing and the mediation of reward- motivational behavior. These various brain regions also give input to the PAG, which via the raphe nuclei in the RVM, and to the locus coeruleus, send descending pain modulatory projections back to the first synapses in the afferent pathways.
Ascending central pathways
The axons of the second order neurons cross the midline and project to the thalamus and a variety of other targets in the brain stem, mid brain and fore brain. Third order neurons, with their cell bodies in the thalamus, project to the primary and secondary somatosensory cortex (S1 and S2), responsible for the sensory-discriminative aspects (intensity, location and quality) of pain, and to limbic cortical areas, such as the anterior cingulate cortex (ACC) and the insular cortex, involved in the mediation of the affective/emotional components (aversiveness) of pain (Figure 1).
Pain matrix
The ascending nociceptive pathways also, directly or indirectly, gives input to other parts of the so called «pain matrix» involved in the emotional and cognitive processing of pain. In addition to the thalamus, the S1 and S2 brain cortex regions and the anterior cingulate and insular cortex, the pain matrix also includes the amygdala, nucleus accumbens, hippocampus and prefrontal cortex as well as the periaqueductal grey (PAG), an important relay station for (among other things) descending pain modulatory pathways (Figure 1). PAG also receives descending input from various cerebral cortical regions, amygdala and hypothalamus. PAG neurons in turn project caudally to the raphe nuclei in the rostral ventral medulla (RVM) and to the locus coeruleus which send descending pain modulatory projections to the brain stem and dorsal horn (Figure 1).
Neurochemistry of pain
The initiation of pain - the first steps in pain signaling
The understanding of the mechanisms by which various stimuli activate the nociceptors, has grown immensely during recent years. One important mile stone was the identification of the transient receptor potential (TRP) channels. This family of ion channels (i. e. «pores» in the cell membranes that regulate the passage of ions and that, at the same time, serve as receptors for chemical substances) has been conserved during evolution and can be found in a vast number of species including yeast, invertebrates and vertebrates, and serve a large range of functions (2). Some members of this family, which are present in nociceptive nerve endings of mammals including man, are activated by thermal stimuli, with different channels being responsible for different temperature ranges, but also by chemical stimuli (3). For example, painful heat (>43°C) activates the TRP vanilloid 1 (TRPV1) channel, that also responds to capsaicin, the pungent ingredient in chili pepper, and protons (i e low pH). Activation leads to the inflow of positive (sodium and calcium) ions and hence depolarization of the cell membrane (Figure 2). Conversely, the TRPM8 receptor responds both to cold temperatures and menthol. These two examples explain the burning sensation after ingestion of spicy («hot») food, and why menthol is experienced as «cold», respectively. In addition, there are a broad range of receptors on these nerve endings that may be activated by various substances that are formed or released in connection to tissue damage and inflammation, such as prostaglandins, nerve growth factor, bradykinin, serotonin, histamine and ATP, (Figure 2). Some of these substances can alter neural excitability directly by activating ion channels on the cell surface, e.g. serotonin, ATP and protons, whereas others, e.g. bradykinin and NGF, mediate their effects through second messengers (4).
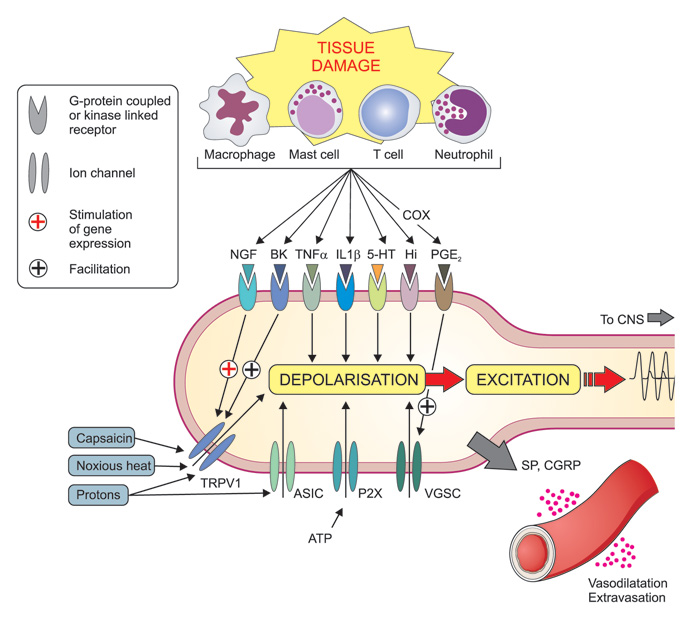
Figure 2. Mechanisms involved in of peripheral sensitization during inflammation. Tissue damage lead to the local formation and release of numerous chemicals around the primary afferent nerve endings due to disruption of cells and activation of various specialized cells including macrophages, mast cells, T-cells, neutrophils and other immune cells. Mediators released include protons (H+), purines (e. g. adenosine triphosphate, ATP), nerve growth factor (NGF), bradykinin (BK), tumor necrosis factor (TNF-?), interleukins (e. g. IL-1?), serotonin (=5-hydroxytrytamine, 5-HT), histamine (Hi) and prostaglandins (e g PGE2). Prostaglandins are formed by cyclooxygenase (COX) enzymes (which are inhibited by non-steroid anti-inflammatory drugs, NSAIDs like e g ibuprofen and naproxen). Together these (and other) mediators form the «inflammatory soup» that contribute to an increased sensitivity of peripheral nociceptors by coupling to membrane-bound ion channels (including transient receptor potential (TRP) channels (e. g. TRPV1), , acid-sensitive ion channels (ASICs) and purinergic (P2X) receptors) and kinase linked receptors (eg NGF-, TNF-? and IL-1? receptors) and G protein-coupled receptors (e. g. bradykinin2- and prostaglandin-receptors). Altogether these mechanisms can lead to depolarization and excitation of the neuron as well as long term alteration of gene regulation (e. g. NGF and bradykinin increasing the expression of TRPV1 - and prostaglandins increasing the expression of voltage gated sodium channels (VGSC). In addition peripheral nociceptor terminals may themselves release substance P (SP) and calcitonin gene related peptide (CGRP), which cause vasodilatation and plasma extravasation (neurogenic inflammation).
Mediators and receptors at the first order synapses
The most important excitatory transmitter substance released by the terminals of primary afferent neurons is glutamate (Figure 3, Table 1). Two types of glutamate receptors are especially important for this signaling. Under normal conditions (to evoke short-lasting «everyday pain»), glutamate activates AMPA receptors on neurons in the dorsal horn and spinal trigeminal complex. In response to prolonged intense nociceptive stimulation, another type of glutamate receptors, the NMDA-receptors are engaged, acting as «turbo aggregates» in these synapses and believed to be involved in sensitization mechanisms (see below). Other proalgesic mediators, released from primary afferent terminals and contributing to sensitization and prolongation of pain (via their own receptors), include substance P (SP), calcitonin gene related peptide (CGRP) and adenosine triphosphate (ATP).
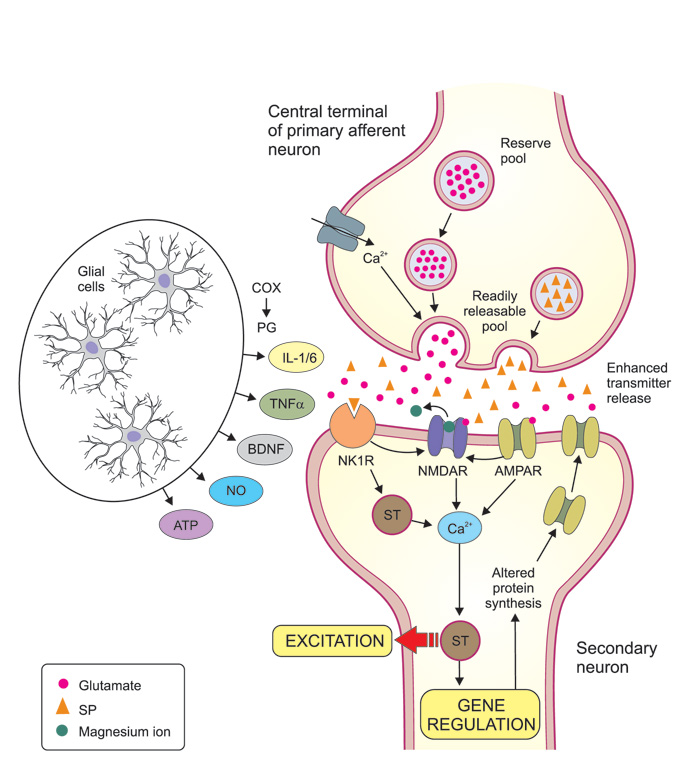
Figure 3. Mechanisms involved in of central sensitization. Central sensitization is largely due to synaptic potentiation between the central terminals of nociceptive primary afferents (top of figure) and secondary neurons in the dorsal horn (or in the trigeminal subnucleus caudalis). Presynaptic mechanisms may include an increased influx of calcium that facilitates transmitter release, and an increase of the releasable transmitter pool. Glutamate and substance P (SP) are both excitatory transmitter substances released by the primary afferents. Glutamate acts on AMPA and NMDA receptors which are both ion channels that directly increase the inflow of calcium into the secondary neurons, while SP acts on neurokinin-1 receptors (NK1R) and increase intracellular calcium via signal transduction (ST) mechanisms. A massive release of glutamate and substance P and subsequent strong stimulation of AMPA and NK1R, will lead to a removal of the magnesium-ion-block that is normally present in the NMDA channel, allowing increased amounts of calcium ions to flow into the postsynaptic cell. Via various ST mechanisms the increased calcium concentration leads to an enhanced excitation of the postsynaptic neuron and long term alterations of gene regulation, which may in turn lead to for example more AMPA receptors in the postsynaptic membrane and thereby enhanced transmission. These mechanisms are triggered and/or facilitated by a large number of extracellular mediator molecules, many of which are released by glial cells (astrocytes and microglia). Ingredients in this «painful soup» includes prostaglandins (formed locally by cyclooxygenases, COX), cytokines like interleukin (IL) 1? and IL 6 and tumor necrosis factor-alpha (TNF-?), brain derived neurotrophic factor (BDNF), nitric oxide (NO) and adenosine tri-phosphate (ATP). Together, all this results in the amplification of pain messages being relayed to higher brain centers. The receptors for these mediator molecules on pre- and postsynaptic neurons are not indicated in figure.
Proalgesic |
· Glutamate (primary afferents and ascending pathways)· Substance P (primary afferents)· Serotonin (descending pathways, effect via excitatory serotonin receptors)· Cholecystokinin (local neurons in dorsal horn and brain) |
Analgesic |
· Opioid peptides (local neurons in dorsal horn and brain)· Noradrenalin (descending pathways)· Serotonin (descending pathways, effect via inhibitory serotonin receptors)· Dopamine (projection neurons in brain)· Gamma amino butyric acid (local neurons in spinal cord) |
It is not fully known, but likely, that the same receptors that regulate function in the peripheral nerve terminals also are functional at the central terminals of the primary afferents. For example, prostaglandin-, bradykinin- and ATP-receptors as well as TRPA1 and calcium channels act to increase transmitter (e g glutamate) release and thus pain. In addition there are presynaptic opioid, GABA, glycine and cannabinoid receptors, which reduce transmitter release from the central nociceptive nerve terminals, and hence, reduce pain (5). Opioid peptides and GABA, released from local interneurons, are likely to be important for the «gate control mechanism", by which non-noxious input is able to suppress pain signaling at the first synapses, even though the regulation has turned out to be more complex than was originally foreseen (6).
Supraspinal pain signaling and pain modulation
Compared to the first order synapses in the afferent nociceptive pathways, less is known about the messenger substances and receptors involved in the continued transmission of pain signals up to (and in) the cerebral cortex, but glutamate and several other neuro messengers are likely to be involved.
For the further processing of nociceptive information in the pain matrix, opioid peptides and the monoamine transmitter dopamine are of obvious importance. The ACC is among the brain regions richest in opioid peptides, and opioid receptors in the ACC (as well as in several other regions) are activated during placebo analgesia. The notion that placebo analgesia is indeed due to an active neuronal mechanism, is illustrated by the finding, almost 40 years ago (7) (and later confirmed see 8), that the analgesic effect of placebo treatment, following e. g. tooth extraction, can be counteracted by pretreatment with the opioid antagonist naloxone. Placebo mechanisms also involve the activation of dopaminergic projections from the ventral tegmental area (VTA) to the nucleus accumbens, ACC and prefrontal cortex (8). These dopamine projections have well-established and crucial importance for reward-motivational behavior in connection to diverse activities such as gambling, jogging, smoking, alcoholism and drug abuse (9, 10).
Descending pain modulation
Another important «output pathway» for the pain matrix is the descending pain modulatory system, with serotonin- and noradrenalin-containing projections from the raphe nuclei and locus coeruleus, respectively (Figure1). Under normal conditions, activations of these pathways inhibit pain signaling already at the first order synapses, either by direct effects or via inhibitory (e. g. opioidergic) interneurons in the dorsal horn and spinal trigeminal nucleus. However, as mentioned above, there is accumulating evidence, that in chronic pain patients, the balance in the pain matrix and in descending pathways may be distorted, possibly by pain itself, or by various precipitating factors (11). Under such conditions there may also be a shift of receptor subtypes, making serotonin and opioids to act as proalgesic substances.
Increased pain sensitivity and chronification of pain
The special clinical problem of chronic pain
Peripheral and central sensitization mechanisms is a major feature when pain and pain hypersensitivity remains, even though peripheral healing is complete after a trauma or a surgical intervention. In most cases the pain successively declines and the threshold for pain returns to pre-injury levels. In a small fraction of patients, however, pain may persist and the acute pain transformed to a chronic pain condition. Chronic pain typically causes a special clinical problem since the pain does not correlate to any peripheral pathology and is often difficult to alleviate. It should emphasized that the mechanisms may differ between acute sensitization and development of chronic pain as well as between different chronic pain conditions.
Peripheral sensitization and the inflammatory soup
Peripheral sensitization is the process whereby the nociceptor due to a change in the chemical milieu reduces its activation threshold and increases the responsiveness. This leads to increased pain sensitivity (hyperalgesia). During tissue trauma inflammatory mediators are released due to disruption of cells, but also by degranulation of mast cells and activation of platelets, macrophages, and immune cells, forming the so-called «inflammatory soup» (Figure 2). By binding to their respective receptors a cascade of events is initiated. Early steps in this process include¬ the activation of multiple intracellular signal transduction pathways in the primary afferent neurons. These include the protein-kinases A (PKA) and C (PKC) and mitogen-activated protein kinases (MAPK) ERK and p38. Activation of these pathways leads to various gene regulations in the cell nuclei of the afferent neurons in the trigeminal and dorsal root ganglia, including increased transcription (production of «RNA-copies") of the genes encoding for TRPs (e. g. TRPV1) and voltage gated sodium channels (VGSC), such as Nav1.7, Nav1.8 and Nav1.9, and to increased synthesis of the channels via translation (protein synthesis using RNA as template). The channels are then transported to the cell membrane of the primary afferents (protein trafficking) where they are phosphorylated, leading to lowered activation threshold of the channels and sensitization of the nociceptor (primary hyperalgesia) (12).
Activation of nociceptive neurons may also lead to release of neuropeptides such as SP and CGRP from their peripheral nerve endings. Both these mediators are strong vasodilators and cause plasma extravasation and may provoke further release of inflammatory mediators from activated platelets, mast cells and immune cells. This neurogenic inflammation enhances sensitization and makes pain worse. (12) (Figure 2).
Central sensitization - sensitization at first order synapses
If a peripheral stimulus is particularly intense, maintained for a prolonged time, and when the peripheral sensory afferent is sensitized, the second order neuron, with its cell body in the spinal dorsal horn/trigeminal subnucleus caudalis, may become sensitized. Even if central sensitization in many ways is comparable to peripheral sensitization, it differs mechanistically. First, second order neurons may receive convergent input not only from nociceptive A? and C-fibers, but also from A? fibers. Therefore, input from A? fibers, such as touch, will be perceived as painful (allodynia) during central sensitization. Second, due to the convergence of many fibers, hyperalgesia will also occur in areas adjacent to the injured site (secondary hyperalgesia). Convergence of superficial as well as deep afferent inputs, from orofacial structures to the second order neurons in the trigeminal complex, may explain our poor ability to localize dull aching pain from deep structures of head and neck (13).
Because central sensitization results in changes within the CNS and thus is not coupled to the presence, intensity or duration of peripheral input, it may also increase pain sensitivity for a prolonged time after the peripheral pathology has disappeared. Thus central sensitization represents an increased amplification of the input signal from the primary afferent neurons (14). In most cases central sensitization successively attenuates when peripheral pathology is resolved.
Clinically central sensitization may be distinguished from peripheral sensitization based on the following observations: 1) pain evoked by innocuous stimuli (allodynia), 2) pain hypersensitivity in areas with no demonstrable pathology, 3) pain that outlasts the stimulus (after sensations), 4) enhanced temporal summation, and 5) the maintenance of pain by low frequency stimuli that normally do not evoke any ongoing pain (15).
Another «spicy soup» around central nerve endings
On the molecular level repeated activation of peripheral afferents increase the release of transmitters such as glutamate and SP at their central terminals with subsequent activation of their respective postsynaptic receptors (Figure 3). Of specific importance for increasing neuronal excitability is the activation of NMDA receptors. Under normal circumstances NMDA receptors are inactivated by a magnesium ion blocking the channel, but sustained release of glutamate may remove this magnesium block and thus activate several intracellular pathways that contribute to the development and maintenance of central sensitization (14). In addition to the critical role of NMDA receptors for central sensitization, other receptors and ion channels are involved. These can separately or together, initiate the activation of multiple intracellular signaling pathways, including PKA and PKC as well as ERK and p38 MAPK (which are also involved in sensitizing mechanisms in primary afferent neurons, see above) that lead to the establishment of increased sensitivity in dorsal horn and trigeminal subnucleus caudalis neurons (13, 14, 16). Furthermore, animal studies indicate a reduction in the spinal level of GABAergic and glycinergic inhibition in neuropathic pain, suggesting that the normal «gate-control» of afferent impulses is out of function (17).
Role of glial cells for central sensitization - not only glue!
It was previously believed that the role of glial cells was to provide structural support to the neurons (glia means glue in Greek). However, during recent years it has become clear that glial cells also play an important role in central sensitization. It is now evident that tissue trauma and inflammation leads to infiltration of macrophages into the spinal dorsal and trigeminal ganglia. This activates satellite glial cells (microglia and astrocytes) which in turn activates ERK and p38 MAPK intracellular pathways. Also release of SP and CGRP from primary afferent neurons modulates satellite glial cell activity. Further, gap junctions ("unnatural» new connections) are established between satellite glial and neuronal cells leading to increased afferent input and enhanced neuronal excitability (13, 18).
In the spinal dorsal horn and trigeminal subnucleus caudalis, activation of microglia and astrocytes cause the release of cytokines that via feedback loops modulate the activity in sensory neurons. Release of cytokines from glial cells further increase AMPA and NMDA receptor activity and thus increase neuronal excitability. Other mediators released from microglia and of importance for central sensitization are chemokines, BDNF, ATP and nitric oxide (13, 14,16) (Figure 3). Interestingly, animal research suggests different underlying mechanisms in glial activation mediating central sensitization in males and females, but further research is needed to establish a possible role for this sex difference in chronic pain states (16).
Importance of the pain matrix for chronification of pain and the overlap between brain regions handling physical and emotional pain
During recent years it has become increasingly evident, not least from studies with brain imaging technique (e. g. functional magnetic resonance imaging, fMRI), that the pain matrix is not only processing physical pain, but also emotional pain, as for example in connection to social exclusion (19). There is also accumulating evidence that severe physical and social pain may alter brain function (20, 21), and that such changes may contribute to the transition from acute to chronic pain (22) and to the comorbidity between chronic pain, depression and anxiety disorders (23). Studies with fMRI on patients with chronic maladaptive pain conditions, e. g. fibromyalgia, show an altered balance between the different components in the pain matrix including the descending pain control from PAG and RVM, allowing much more nerve activity to pass up-streams to the pain matrix (11). Recent brain imaging studies also indicate that patients with trigeminal neuropathic pain or temporomandibular joint disorders have functional changes in multiple brain regions, suggesting a role for emotional and cognitive modulation as well as reward processing in orofacial pain (24). These findings not only ads complexity to the previously described pain amplifying mechanisms at nociceptor level in the periphery and in the dorsal horn of the spinal cord/trigeminal brainstem sensory nuclear complex, but may also open new avenues for treatment of chronic pain (20). It also emphasizes the importance of the clinician's attitude and skills when pain is present or to be expected (see clinical box). Thus, basic pain mechanisms are influenced by factors as fear, expectation and the clinical handling and atmosphere (8).
Conclusion
The knowledge about the neurochemical circuitry involved in peripheral, spinal and supraspinal pain processing has increased enormously during recent years, but is even though still incomplete. Obviously a large number of both facilitating and inhibitory mechanisms interact in a most intriguing and complicated way. With increased knowledge within this field, new targets for treatment are likely to emerge, that may not only permit the immediate counteraction of pain, but also make it possible to prevent the development of chronic pain.
Acknowledgements
The drawing of the figures by Scientific ilustrator Sole Lätti is gratefully acknowledged. The authors declare that they have no competing financial interests.
References
Zaman J, Vlaeyen JW, Van Oudenhove L, Wiech K, Van Diest I. Associative fear learning and perceptual discrimination: A perceptual pathway in the development of chronic pain. Neurosci Biobehav Rev. 2015 Apr; 51: 118 - 125.
Nilius B, Owsianik G. The transient receptor potential family of ion channels. Genome Biol. 2011; 12(3): 218.
Moran MM, McAlexander MA, Szallasi A. Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov. 2011 Aug 1; 10(8): 601 - 20.
Julius D, Basbaum AI, Molecular mechanisms of nociception. Nature 2001; 14: 203 - 10.
Woolf CJ, Ma Q. Nociceptors-noxious stimulus detectors. Neuron 2007; 55: 353 - 64.
Price TJ1, Cervero F, Gold MS, Hammond DL, Prescott SA. Chloride regulation in the pain pathway. Brain Res Rev. 2009 Apr; 60(1): 149 - 70.
Levine JD, Gordon NC, Fields HL. The mechanism of placebo analgesia. Lancet. 1978 Sep 23; 2(8091): 654 - 7.
Benedetti F. Placebo and the new physiology of the doctor-patient relationship. Physiol Rev. 2013 Jul; 93(3): 1207 - 46.
Leknes S1, Tracey I. A common neurobiology for pain and pleasure. Nat Rev Neurosci. 2008 Apr; 9(4): 314 - 20.
Narita M, Matsushima Y, Niikura K, Narita M, Takagi S, Nakahara K, Kurahashi K, Abe M, Saeki M, Asato M, Imai S, Ikeda K, Kuzumaki N, Suzuki T. Implication of dopaminergic projection from the ventral tegmental area to the anterior cingulate cortex in ?-opioid-induced place preference. Addict Biol. 2010 Oct; 15(4): 434 - 47.
Ossipov MH, Morimura K, Porreca F. Descending pain modulation and chronification of pain. Curr Opin Support Palliat Care. 2014 Jun; 8(2): 143 - 51.
Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009; 139: 267 - 84.
Sessle, B J. Peripheral and central mechanisms of orofacial inflammatory pain. Int Rev Neurobiol. 2011 97: 179 - 206
Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009; 10: 895 - 926.
Woolf CJ Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011 Mar; 152(3 Suppl): S2 - 15.
Mifflin KA, Kerr BJ. The transition from acute to chronic pain: understanding how different biological systems interact. Can J Anesth/J Can Anesth. 2014; 61: 112 - 22.
Castro-Lopes JM, Tavares I, Coimbra A. GABA decreases in the spinal cord dorsal horn after peripheral neurectomy. Brain Res. 1993; 620(2): 287 - 91.
Verma V, Sheikh Z, Ahmed AS. Nociception and role of immune system in pain. Acta Neurol Belg. 2014, DOI 10.1007/s13760 - 014 - 0411-y.
Eisenberger NI. The pain of social disconnection: examining the shared neural underpinnings of physical and social pain. Nat Rev Neurosci. 2012 May 3; 13(6): 421 - 34.
Saab CY.Pain-related changes in the brain: diagnostic and therapeutic potentials. Trends Neurosci. 2012 Oct; 35(10): 629 - 37.
van Harmelen AL, Hauber K, Gunther Moor B, Spinhoven P, Boon AE, Crone EA, Elzinga BM. Childhood emotional maltreatment severity is associated with dorsal medial prefrontal cortex responsivity to social exclusion in young adults. PLoS One. 2014 Jan 8; 9(1): e85107.
Apkarian AV1, Baliki MN, Farmer MA. Predicting transition to chronic pain. Curr Opin Neurol. 2013 Aug; 26(4): 360 - 7.
Navratilova E1, Porreca F1. Reward and motivation in pain and pain relief. Nat Neurosci. 2014 Oct; 17(10): 1304 - 12.
Lin CS. Brain signature of chronic orofacial pain: a systematic review and meta-analysis on neuroimaging research of trigeminal neuropathic pain and temporomandibular joint disorders. Plos One. 2014 Apr 23; 9 (4): e94300: 1 - 13.
Professor. Department of Physiology and Pharmacology, Karolinska Institutet, Stockholm, Sweden
DDS, PhD, Professor and Senior Consultant. Department of Dental Medicine and Scandinavian Center for Orofacial Neurosciences (SCON), Karolinska Institutet, Stockholm, Sweden
Professor Emeritus. Department of Physiology and Pharmacology, Karolinska Institutet, Stockholm, Sweden
Corresponding author: Ernst Brodin, e-mail: ernst.brodin@ki.se
This paper has been peer reviewed.
Brodin E, Ernberg M, Olgart L. Nor Tannlegeforening Tid. 2016; 126: 28-33 Neurobiology: General considerations - from acute to chronic pain.