Osteogenesis imperfecta - odontologiske implikationer
Osteogenesis imperfecta (OI) er en arvelig, sjælden bindevævssygdom, der forårsager medfødt knogleskørhed og øget risiko for knoglebrud samt en række andre symptomer relateret til bindevævet, herunder risiko for dentinogenesis imperfecta (DI). Diagnosen DI stilles på baggrund af kliniske fund og røntgenologiske karakteristika forårsaget af defekt dentin. DI kan forekomme isoleret eller som en del af OI.
Diagnosen OI stilles klinisk, men både OI og DI kan bekræftes med genetiske analyser. Opfølgning og behandling af OI og af DI finder sted på specialiserede centre med fokus på særlige forhold omkring disse og andre sjældne diagnoser.
I denne artikel beskrives udredning, opfølgning og behandling af DI og OI i et felt, hvor medicinen og odontologien krydser veje, hvilket indbyder til øget opmærksomhed og samarbejde.
Klinisk relevans | |
|---|---|
Den odontologiske diagnose dentinogenesis imperfecta (DI) kan forekomme isoleret eller være en del af en generel bindevævssygdom associeret til et skrøbeligt skelet, osteogenesis imperfecta (OI). I denne artikel beskrives udredning, opfølgning og behandling af DI og OI i et felt, hvor medicinen og odontologien krydser veje, hvilket indbyder til øget opmærksomhed og samarbejde. |
Osteogenesis imperfecta (OI) er en arvelig bindevævssygdom, der inddeles i forskellige undertyper med forskellig sværhedsgrad relateret til specifikke forandringer i og omkring kollagen type 1-proteinet, der er et af de hyppigst forekommende proteiner i kroppen (1). Symptomerne relaterer sig til flere organer og omfatter knoglefrakturer, knogledeformiteter, lav knoglemineraldensitet (osteoporose), lav højde, abnorm tanddannelse, malokklusion, tidligt høretab, blå sclerae og hypermobilitet. De kognitive og sociale funktioner er normale. Sygdommen manifesterer sig ofte tidligt i livet, hvor debutsymptomet kan være en skeletal fraktur opstået ved fødslen, eller når barnet er i færd med at lære at gå. Sværhedsgraden af OI er meget varierende fra mild og næsten asymptomatisk sygdom til svær skeletal dysplasi og død tidligt i livet (figur 1). Langt de fleste tilfælde er milde.
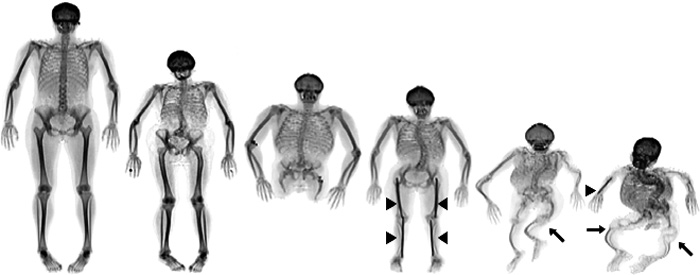
Figur 1. OI manifesterer sig i skelettet betydeligt varierende fra næsten asymptomatisk, mild OI til svær OI med deforme knogler. Røntgenbilleder af voksne med OI. (Billede fra internettet).
OI forårsages af mutationer i generne, der koder for proteinet kollagen type 1 eller i gener af betydning for dannelsen af kollagen type 1 (2). Mutationerne nedarves helt overvejende efter et dominant nedarvningsmønster, men et recessivt mønster ses specielt i familier, der er indgifte. I omkring halvdelen af tilfældene er der tale om nymutationer, og forældrene er således raske. Den sygdomsfremkaldende mutation forårsager enten en kvalitativ defekt i kollagen type 1-proteinet, hvilket bryder den regelmæssige struktur i bindevævets opbygning og derfor ofte resulterer i sværere sygdom, eller mutationen forårsager en kvantitativ defekt med dannelse af et normalt protein, men i en reduceret mængde, hvilket resulterer i mildere sygdom (3).
Den samlede prævalens af OI er estimeret til 11 pr. 100.000 (4), hvilket rubricerer OI som en sjælden sygdom. Klassisk skelner man imellem fire kliniske undertyper (I-IV) som beskrevet af David Sillence i 1979 (5). Gennem de seneste 10 - 15 år er der beskrevet yderligere 11 typer, som klinisk kun afviger beskedent fra de oprindeligt beskrevne typer, men genetisk forårsages af forandringer i andre gener (6).
Symptomer ved osteogenesis imperfecta
Der er stor variabilitet i antal af knoglefrakturer selv indenfor samme familie, og en barndom med OI kan være præget af alt fra meget få til multiple knoglefrakturer. De første knoglefrakturer kan allerede debutere intrauterint eller ved fødslen, men oftest sker det først omkring den alder, hvor barnet lærer at gå. Knoglefraktur kan ses i samtlige knogler, men involverer oftest rørknoglerne. Hyppigheden af knoglefrakturer aftager som regel i de sene teenageår og i den tidlige voksenalder for dernæst at vende tilbage i forbindelse med aldersbetinget knogleskørhed. Knogledeformiteter, fx buede rørknogler eller skæv ryg (skoliose), kan være betydende (figur 1). Hypermobilitet skyldes eftergivelighed i ledbånd og ledkapsler og kan ved både mild og svær OI være medvirkende årsager til, at nogle af børnene opnår de motoriske milepæle senere end normalt, og at en andel af både børn og voksne har motoriske funktionsbegrænsninger med behov for hjælpemidler.
Legemshøjden hos den voksne med OI kan være normal eller betydelig reduceret, i de sværeste tilfælde til under 100 cm.
Blåfarvning af øjets sclerae ses hos en høj andel af patienterne, men kan også forekomme hos mange raske spædbørn. Et tidligt høretab ses hos 50 % og kan debutere allerede i teenageårene. Årsagerne til høretab kan bl.a. være frakturer/deformiteter af de små knogler i mellemøret eller ændret form af kraniet.
Odontologiske karakteristika ved osteogenesis imperfecta
DI kan ses i både det primære og det permanente tandsæt ved OI. DI karakteriseres ved hvidlig til grålig-blåligt farvede tænder, som kan ændre sig til gullig-brunlige farver fx i forbindelse med tab af emalje (figur 2). Tilstanden indebærer ofte affrakturering af emalje og en efterfølgende attrition, der kan antage ekstreme former. Denne attrition er oftest mest udtalt i det primære tandsæt (figur 2). I det permanente tandsæt kan der optræde egentlige kronefrakturer grundet den anomale dentinstruktur. Røntgenologiske karakteristika er korte rødder med obliteration af pulpa samt løgformede tandkroner med cervical konstriktion (figur 2 og 3). Yderligere er det ofte karakteristisk med mandibulært overbid og åbent bid (figur 3 og 4) (7 - 9).
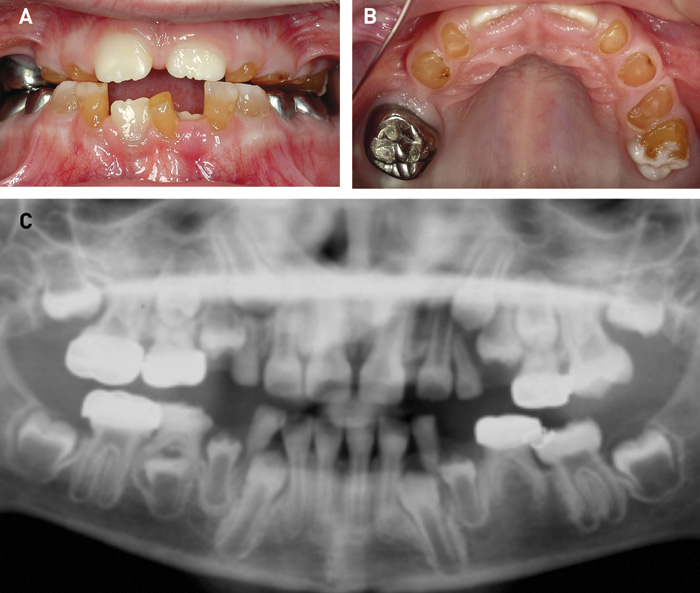
Figur 2. Klinisk manifestation af DI hos et barn på syv år med OI. A, B. Afskalning af emalje og udtalt attrition på de primære tænder. Første permanente molar (26) er ligeledes med afskalning af emalje og eksponering af den gule dentin. Stålkronen på 26 er mistet. C. Røntgenbilledet viser meget store pulpacavae på de umodne permanente tænder (fx præmolarer og andenmolarer), mens de primære tænder er med pulpaobliteration.
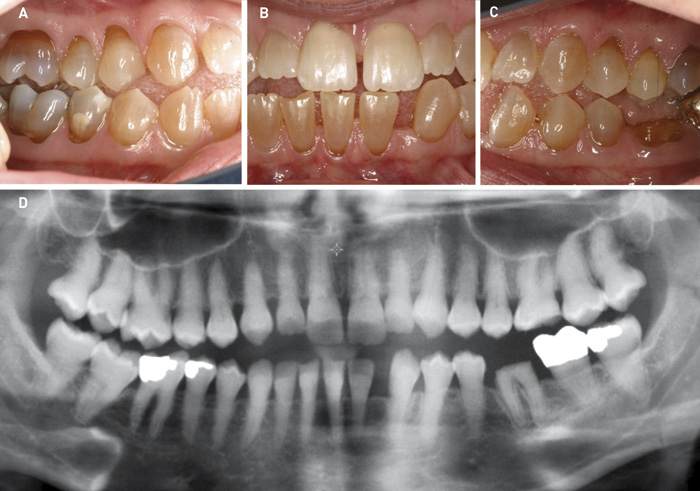
Figur 3. Klinisk manifestation af DI hos kvinde på 36 år med OI type 4. A, B, C. Gråbrun eller gulligbrun farve på kronerne. Finerkrone på anden permanente molar i venstre side af underkæben (37) og mistet krone ses på første permanente molar i venstre side af underkæben (36). Derudover ses asymmetrisk okklusion med et lille underbid og åbent bid. D. Røntgenoptagelse i form af panoramaoptagelse viser, at alle 32 permamente tænder er til stede. Bemærk pulpaobliteration.
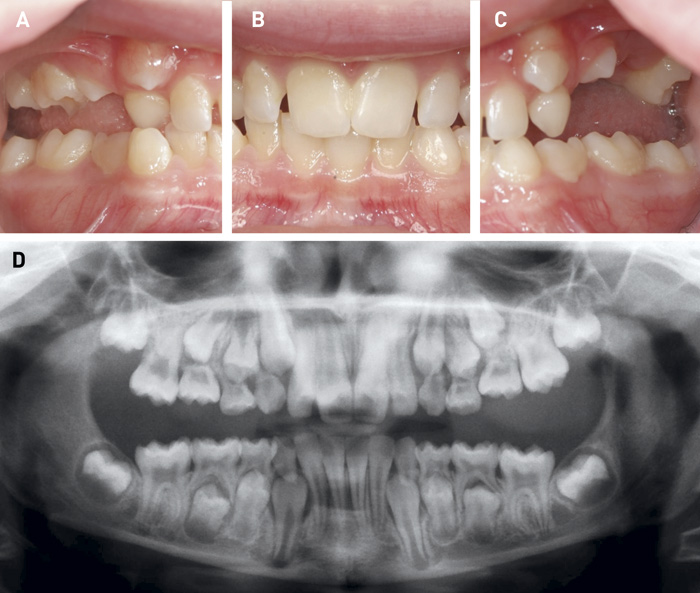
Figur 4. Barn på 11 år med svær OI og uden umiddelbare tegn på DI. A, B, C. Atypisk eruption af tænder i overkæben. Bilateralt åbent bid. D. Panoramarøntgen viser normal aftegning af pulpa i både primære og permanente tænder.
Dentindefekterne har samme histopatologiske baggrund som ved OI, dvs. enten kvalitative eller kvantitative afvigelser i kollagen (10). Det må antages, at de kvalitative afvigelser (OI type 3 og OI type 4) giver de sværeste dentale manifestationer.
Forekomsten af DI hos OI-patienter er rapporteret til mellem 19 - 42 % afhængigt af fx diagnostiske kriterier og forskelle i studiepopulationerne (11,12) (tabel 1). Der er tale om tværsnitsstudier, hvor den generelle usikkerhed er stor, hvilket generelt knytter sig til undersøgelse af prævalens ved sjældne tilstande. Milde tilfælde kan underdiagnosticeres, og svære tilfælde kan have vanskeligt ved at møde op til undersøgelse.
Isoleret dentinogenesis imperfecta
DI kan også optræde som en isoleret dental anomali. Traditionelt inddeles DI efter Shields klassifikation (1973), som beskriver tre forskellige typer (DI-I, DI-II og DI-III) (13) baseret på kliniske symptomer. Af disse er kun DI-I betinget af kollagendefekter og dermed relateret til OI. La Dure-Molla et al. (14) har foreslået en DI-klassifikation baseret på det faktum, at DI-II & DI-III (Brandy wine type) i lighed med dentindysplasi type II (DD-II) er forårsaget af mutationer i genet, der koder for dentin sialophosphoprotein (DSPP), som ligger på kromosom 4q (11). Disse typer repræsenterer forskellige fænotyper med forskellig klinisk sværhedsgrad. Dentin sialophosphoprotein tilhører gruppen af proteiner i den ikke-kollagenholdige del af dentinens matrix. Det er således af særlig differentialdiagnostisk betydning i forbindelse med omtale af OI, at DI også findes som en isoleret dental anomali (DI type II), hvor det kliniske billede af tænderne ligner billedet ved den OI-relaterede DI (DI-type I).
Forekomsten af isoleret DI er rapporteret til 1: 8000 (15) baseret på Witkops tal fra 1957 (16).
Diagnostik og den genetiske årsag til osteogenesis imperfecta/dentinogenesis imperfecta
Diagnosen OI stilles klinisk af læger med specialviden om sygdommen og baseres på de fænotypiske træk og anamnesen. En positiv familiehistorie kan være af diagnostisk betydning. Ofte stilles diagnosen i barndommen, men mild OI kan gå udiagnosticeret hen. Der er eksempler på, at diagnosen DI stilles en årrække før diagnosen OI.
Hos omkring 90 % med klinisk verificeret OI findes forandringer i et af de to gener, COL1A1 og COL1A2, der koder for kollagen type 1-proteinet (15). I takt med at molekylærbiologiske metoder har vundet frem og tilbydes rutinemæssigt, har flere forsøgt at karakterisere og kategorisere patienter med OI. Dertil kommer, at mutationsundersøgelser er blevet billigere og dermed mere tilgængelige og kan bekræfte og supplere den initiale diagnostik af OI. Ved en simpel hudprøve kan kollagen fra hudens fibroblaster undersøges ved gelelektroforese og bidrage til sygdomskarakteristikken, omend undersøgelsen ikke rutinemæssigt bruges i dag grundet øget tilgængelighed af genetiske analyser. En klinisk mistanke om OI kan således suppleres med undersøgelse af, hvorvidt der er en kvalitativ eller kvantitativ defekt kollagen type 1, og en undersøgelse for mutationer i gener associeret med OI. Undersøgelser har vist, at patienter med OI og samtidig DI ofte har en kvalitativ defekt i kollagen type 1-proteinet. En nylig dansk undersøgelse har dog vist, at OI/DI også konstateres hos personer med kvantitativt defekt protein (ikke-publicerede data).
Odontologisk behandling ved osteogenesis imperfecta
Indikation for behandling af DI kan være såvel funktionel som psykosocial og/eller kosmetisk. Målet med en intervention er at bevare et funktionsdygtigt tandsæt med en okklusion, der for den enkelte er acceptabel. Ofte har disse patienter selv efter behandling væsentlige afvigelser fra det, der kaldes neutral okklusion og normale vertikale og horisontale kæberelationer. Hvad angår kosmetiske og psykosociale forhold kan mere eller mindre omfattende protetisk behandling være aktuel.
I børne- og ungepopulationen er det ofte nødvendigt at gennemføre en del intermediære behandlinger, der har til hensigt at beskytte det skrøbelige tandmateriale, at sikre normal eruption og at tilstræbe en harmonisk okklusionsudvikling. Disse behandlinger kan omfatte plastopbygning af tandkroner, påsætning af stålkroner, tiltag mhp. fastholdelse af bidhøjde samt interceptiv ortodonti. I den permanente dentition kan elementer som fremstilling af attritionshæmmende skinne være aktuel. Hos den unge voksne kan mere permanent protetisk behandling komme på tale, fx i form af guldkapper, kroner og/eller broer for at skabe et æstetisk og funktionelt acceptabelt tandsæt. Dette er aktuelt, når der forekommer kronefrakturer og/eller æstetisk kompromitterende farveafvigelser. Kronebehandling på samtlige DI-tænder med det formål at beskytte de naturlige tænder er ikke en hensigtsmæssig behandlingsstrategi. Det er nemlig erfaringen, at der med tiden kan optræde collumfrakturer og dermed tab af hele den kliniske krone, uanset eventuel kronebehandling (figur 5).
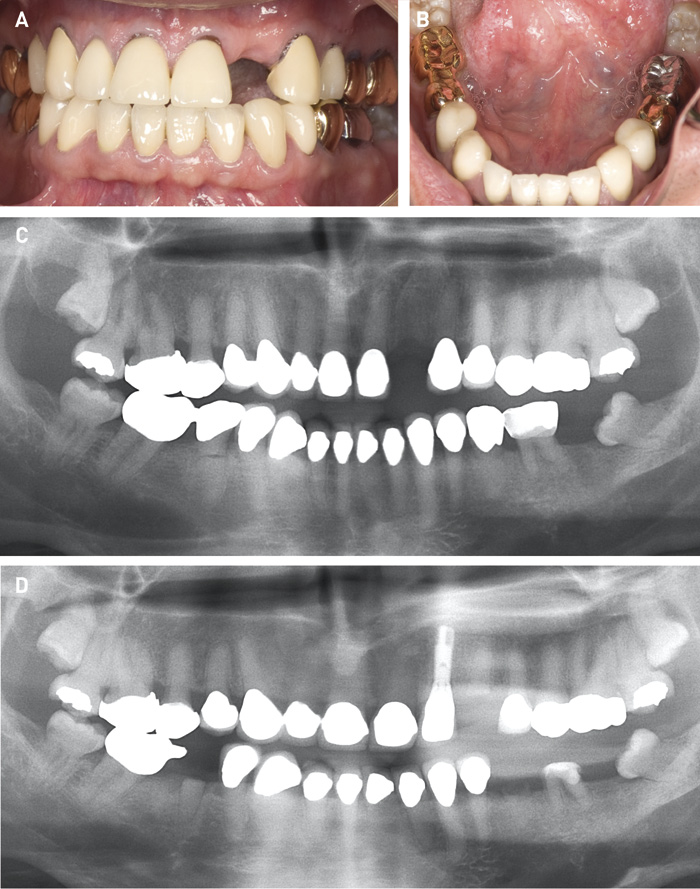
Figur 5. A, B. Fuldkroner på alle tænder fraset fire molarer. 22 er ekstraheret efter tidligere kronebehandling og efterfølgende kronefraktur. C. Panoramarøntgen (45-årig), der viser generel pulpaobliteration, retention af 18 og 28, fravær af 22 og 37 og rodfraktur 44. D. Panoramarøntgen fem år senere. Implantat med krone på 22. Yderligere tre tænder med cervikal rodfraktur og tab af den kliniske krone (23, 36, 35).
Mulighederne for endodontisk behandling af DI-tænder er begrænsede og må anses for at være en specialiseret behandlingsopgave. Tanderstatninger i form af implantatbaseret protetik synes at være en mulighed for OI-patienten med dette behov. Uanset at mange patienter med OI modtager behandling med lavdosis bisfosfonat, er der ikke rapporteret tilfælde af medicininduceret osteonekrose i forbindelse med tandekstraktioner eller anden dento-alveolær kirurgi på denne patientgruppe (17 - 19).
Kæbefrakturer hos patienter med OI kan givetvis forekomme, men er kun i meget begrænset omfang rapporteret i faglitteraturen (20,21).
I forbindelse med behandling af patienter med DI er det almindelig kendt, at tidlig start på behandlingen anbefales. Behandlingerne er tidskrævende og ofte vanskelige at gennemføre. Hyppig og regelmæssig kontrol er påkrævet, og en langsigtet behandlingsplan bør ligge til grund for tandplejens behandlingstilbud. Behandlingsplanlægningen sker i et samarbejde mellem den lokale tandlæge og et af de to odontologiske videnscentre på hhv. Rigshospitalet og Aarhus Universitetshospital. De Odontologiske Videnscentre tilbyder rådgivning til såvel patienterne selv som til de tandlæger, der har kontakt til patienter med OI. Derudover tilbyder videncentrene specialiseret behandling, når dette er påkrævet. Omfattende stålkronebehandling, protetisk behandling og endodontisk behandling ved DI er eksempler på specialiseret behandling i videnscenterregi. Implantatbehandling og visse former for ortodontisk behandling på patienter med OI med eller uden DI er også eksempler på det specialiserede behandlingstilbud i de odontologiske videnscentre.
Vigtigt at vide for tandlægen
Ved OI er der risiko for basilar impression eller instabilitet af de øverste cervikalhvirvler, især hos patienter med svære sygdomsmanifestationer. Derfor kan det være nødvendigt at være varsom ved lejring og manipulation af hoved og hals. Ofte vil patienten selv vide, om det er en relevant problemstilling.
Ved OI er der en øget risiko for knoglefrakturer. Derfor skal fx tandekstraktioner udføres med agtpågivenhed og med skånsom teknik.
Medicinsk, kirurgisk og terapeutisk behandling ved osteogenesis imperfecta
I erkendelse af at der generelt kan være stor forskel i sjældne sygdommes sværhedsgrad, kompleksitet, forløb over tid og behov for kontakt og indsats, er der i Danmark oprettet to højtspecialiserede Centre for Sjældne Sygdomme på henholdsvis Aarhus Universitetshospital (CSS-AUH) og Rigshospitalet (CSS-RH) (22). Dette tiltag sker også i lyset af, at fagpersoners kendskab til den enkelte sygdom og følgerne heraf ofte netop pga. sjældenheden vil være mangelfuldt, således at der er en særlig risiko for, at diagnostik, behandling, rehabilitering og opfølgning ikke bliver varetaget tilstrækkeligt fagligt kvalificeret og rettidigt. Indsatsen varetages i et tæt samarbejde mellem relevante specialer, som bl.a. omfatter pædiatri, medicinsk endokrinologi, ortopædkirurgi, klinisk genetik og audiologi i samarbejde med odontologi. Der er løbende samarbejde med patientens lokale sundhedsvæsen, fysioterapeuter og hjemkommune. Patientens praktiserende læge varetager de almenmedicinske behov, mens undersøgelse og behandling i centrene specifikt er rettet mod OI (23).
Patienter mistænkt for OI kan henvises til udredning på centrene, og alle patienter med OI bør henvises (22). Hyppigheden af besøg planlægges ud fra en individuel vurdering af behov og sygdommens sværhedsgrad.
OI hører under en højt specialiseret funktion, og i henhold til Sundhedsstyrelsens gældende specialeplan er denne placeret på CSS-AUH og CSS-RH. Tillige kan Osteoporoseklinikkerne på Hvidovre Hospital og Odense Universitetshospital varetage osteoporosebehandlingen. På CSS-AUH er tilbuddet organiseret således, at børn op til 16 - 18 år er tilknyttet selve centret, hvorimod voksne er tilknyttet Medicinsk Endokrinologisk Afdeling, MEA, hvad angår kontrol, behandling og koordination mellem involverede specialer. På CSS-RH er alle aldersklasser tilknyttet centret, mens osteoporosebehandlingen af de voksne varetages af Osteoporoseklinikken på Hvidovre Hospital.
Den medicinske behandling har til formål at opbygge og styrke knoglerne for at forebygge knoglefrakturer, deformiteter og smerter. Patienten skal sikres nødvendig tilførsel af kalk og D-vitamin, der begge er væsentlige for det skeletale helbred. I tilfælde, hvor egentlig medicinsk behandling af osteoporose skønnes hensigtsmæssigt, benyttes medicin af typen bisfosfonat. Da patienterne er relativt få i antal, er der kun ganske få velgennemførte videnskabelige undersøgelser, som dokumenterer effekten af den medicinske behandling. I undersøgelserne er fundet god effekt af bisfosfonat på lindring af knoglesmerter, men begrænset evidens for forebyggelse af brud og deformiteter, særligt for den voksne patientgruppe (24).
Ortopædkirurgerne varetager frakturbehandling og korrektion af deformiteter. Behandlingerne omfatter bandagering, tilpasning af skinner og korset samt knoglestøttende og deformitetskorrigerende kirurgisk behandling bl.a. med indsættelse af marvsøm. Endvidere foretages løbende i samarbejde med fysioterapeut vurdering af behov for hjælpemidler.
Mange patienter med OI har funktionshæmning bl.a. pga. deformiteter. Derfor er fysioterapeuter og ergoterapeuter en nødvendig del af det samlede behandlingstilbud. Det varetages mest hensigtsmæssigt via patientens hjemkommune.
Aktuelle studier og danske osteogenesis imperfecta-projekter
Ganske få studier har undersøgt sammenhængen mellem OI og DI. Få studier har sammenlignet sværhedsgraden af OI med tilstedeværelsen af DI og fundet den højeste prævalens af DI blandt personer med betydelig skeletdeformitet og dermed svær OI (25). Generelt har alle studier fundet meget varierende præsentation af DI. Historisk har personer med mild OI været opdelt i undertyper baseret på, om de har DI eller ej. Dette har ikke klinisk betydning for den enkelte patient.
Gennem de seneste fem år har et tværfagligt samarbejde fundet sted omkring forskning indenfor OI. Håbet er, at dette samarbejde vil forplante sig til det tværfaglige kliniske samarbejde indenfor OI. Mere specifikt har et tværsnitsstudie for nylig resulteret i et nyt samarbejde mellem MEA, AUH og Tandlægehøjskolen, Aarhus Universitet, hvor 78 voksne med OI er undersøgt for samtidig DI (ikke-publicerede data). Aktuelt foregår der herhjemme desuden en række andre undersøgelser af knogle, fænotype, dødelighed, hørelse samt medicinsk behandling af patienter med OI.
Osteogenesis imperfecta symptomer | |
|---|---|
Dentogenesis imperfecta, lav højde, relativt stort kranium, deforme knogler, skoliose, mange brud, blå sclerae, positiv familiehistorie |
Sammenfatning
Den overordnede og koordinerende behandling af patienter med OI og DI varetages af CSS-AUH og CSS-RH i samarbejde med lokalt sundhedsvæsen og på odontologiske videnscentre i samarbejde med lokale tandlæger i kommunal tandpleje eller privat praksis. Der kan på ethvert tidspunkt henvises til specialvurderinger på centrene herunder enten Odontologisk Videnscenter eller Center for Sjældne Sygdomme i Aarhus og på Rigshospitalet, der også samarbejder på tværs af faggrænser og geografi.
Et tilbud om en tværfaglig højt specialiseret funktion er nødvendigt for mange sjældne sygdomme, heriblandt OI og DI, men fx også Marfan syndrom og ektodermale dysplasier. Med en sådan organisation kan den nødvendige viden, ekspertise og kvalitet til gavn for patienterne sikres. Det at samle patienterne få steder danner endvidere det bedste grundlag for at erhverve ny viden.
I det daglige kliniske arbejde er det vigtigt for tandlæger at være bekendt med, at DI kan ses i kombination med udtalt skeletal skrøbelighed og være led i en generel bindevævssygdom som OI. I faktaboksen er angivet de symptomer og fund, som kan få tandlægen til at overveje diagnosen.
Konsekvensen af, at DI ses ved alle sværhedsgrader af OI, bør komme patienten til gode. Det anbefales, at alle patienter med OI tilbydes mindst én højt specialiseret odontologisk undersøgelse, uafhængigt af om der er kliniske symptomer på DI eller ej. Endvidere anbefales det at henvise patienter mistænkt for eller med bekræftet OI til et af sjældnecentrene.
English summary
Hald JD, Farholt S, Gjørup H, Haubek D.
Osteogenesis imperfecta - oral implications
690-7
Osteogenesis imperfecta (OI) is an inherited, rare connective tissue disorder leading to bone fragility and hence an increased risk of bone fractures and a number of other symptoms related to the connective tissue, including risk of dentinogenesis imperfecta (DI). The diagnosis DI is based on clinical findings and radiographic characteristics as a consequence of defective dentine. DI can appear as isolated DI or as a part of OI. OI is a clinical diagnosis, however both OI and DI can be confirmed by genetic analyses.
Follow-up and treatment of OI and DI take place at specialized centers with special knowledge and expertise on rare inherited disorders.
This article describes investigations, follow-up and treatment of DI and OI in a field where special attention and increased cooperation between medical physicians and dentists are of major importance.
Litteratur
Byers PH, Steiner RD. Osteogenesis imperfecta. Annu Rev Med 1992; 43: 269 - 82.
Byers PH, Wallis GA, Willing MC. Osteogenesis imperfecta: translation of mutation to phenotype. J Med Genet 1991; 28: 433 - 42.
Lund AM. Biokemiske og molekylærgenetiske studier ved osteogenesis imperfecta. Ugeskr læger 2002; 164.
NCBI. GeneReviews. Internet Communication. (Set januar 2016). Tilgængelig fra: URL: http: //www.ncbi.nlm.nih.gov/books/NBK1295/
Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 1979; 16: 101 - 16.
Marini JC, Reich A, Smith SM. Osteogenesis imperfecta due to mutations in non-collagenous genes: lessons in the biology of bone formation. Curr Opin Pediatr 2014; 26: 500 - 7.
Jensen BL, Lund AM. Osteogenesis imperfecta: clinical, cephalometric, and biochemical investigations of OI types I, III, and IV. J Craniofac Genet Dev Biol 1997; 17: 121 - 32.
Rizkallah J, Schwartz S, Rauch F et al. Evaluation of the severity of malocclusions in children affected by osteogenesis imperfecta with the peer assessment rating and discrepancy indexes. Am J Orthod Dentofacial Orthop 2013; 143: 336 - 41.
Stenvik A, Larheim TA, Storhaug K. Incisor and jaw relationship in 27 persons with osteogenesis imperfecta. Scand J Dent Res 1985; 93: 56 - 60.
Lund AM, Jensen BL, Nielsen LA et al. Dental manifestations of osteogenesis imperfecta and abnormalities of collagen I metabolism. J Craniofac Genet Dev Biol 1998; 18: 30 - 7.
Malmgren B, Norgren S. Dental aberrations in children and adolescents with osteogenesis imperfecta. Acta Odontol Scand 2002; 60: 65 - 71.
Saeves R, Lande WL, Ambjørnsen E et al. Oral findings in adults with osteogenesis imperfecta. Spec Care Dentist 2009; 29: 102 - 8.
Shields ED, Bixler D, el-Kafrawy AM. A proposed classification for heritable human dentine defects with a description of a new entity. Arch Oral Biol 1973; 18: 543 - 53.
de La Dure-Molla M, Philippe FB, Berdal A. Isolated dentinogenesis imperfecta and dentin dysplasia: revision of the classification. Eur J Hum Genet 2015; 23: 445 - 51.
Barron MJ, McDonnell ST, Mackie I et al. Hereditary dentine disorders: dentinogenesis imperfecta and dentine dysplasia. Orphanet J Rare Dis 2008; 20; 3: 31. doi: 10.1186/1750 - 1172 - 3 - 31.
Witkop CJ. Hereditary defects in enamel and dentin. Acta Genet Stat Med 1957; 7: 236 - 9.
Christou J, Johnson AR, Hodgson TA. Bisphosphonate-related osteonecrosis of the jaws and its relevance to children - a review. Int J Paediatr Dent 2013; 23: 330 - 7.
Schwartz S, Joseph C, Iera D et al. Bisphosphonates, osteonecrosis, osteogenesis imperfecta and dental extractions: a case series. J Can Dent Assoc 2008; 74: 537 - 42.
Hennedige AA, Jayasinghe J, Khajeh J et al. Systematic review on the incidence of bisphosphonate related osteonecrosis of the jaw in children diagnosed with osteogenesis imperfecta. J Oral Maxillofac Res 2014; 4: e1.
Feifel H. The surgical treatment of mandibular fractures in a child with osteogenesis imperfecta. Int J Oral Maxillofac Surg 1996; 25: 360 - 2.
Gallego L, Junquera L, Pelaz A et al. Pathological mandibular fracture after simple molar extraction in a patient with osteogenesis imperfecta treated with alendronate. Med Oral Patol Oral Cir Bucal 2010; 15: e895 - 7
SUNDHEDSSTYRELSEN. Specialeplanen. Specialeplanen 2010. (Set januar 2016). Tilgængelig fra: URL: https://sundhedsstyrelsen.dk/da/sundhed/planlaegning-og-beredskab/specialeplanlaegning
SUNDHEDSSTYRELSEN. National Strategi for Sjældne Sygdomme 2014. (Set januar 2016). Tilgængelig fra: URL: https://sundhedsstyrelsen.dk/da/udgivelser/2014/national-strategi-for-sjaeldne-sygdomme
Hald JD, Evangelou E, Langdahl BL et al. Bisphosphonates for the Prevention of Fractures in Osteogenesis Imperfecta: Meta-Analysis of Placebo-Controlled Trials. J Bone Miner Res 2015; 30: 929 - 33.
Lukinmaa PL, Ranta H, Ranta K et al. Dental findings in osteogenesis imperfecta: I. Occurrence and expression of type I dentinogenesis imperfecta. J Craniofac Genet Dev Biol 1987; 7: 115 - 25.
læge, ph.d., Medicinsk Endokrinologisk Afdeling, MEA, Aarhus Universitetshospital
overlæge, ph.d., Center for Sjældne Sygdomme, Børneafdeling A, Aarhus Universitetshospital
overtandlæge, ph.d., Odontologisk Landsdels- og Videnscenter, Kæbekirurgisk afdeling, Aarhus Universitetshospital
professor, dr.odont, ph.d., Sektion for Pædodonti, Institut for Odontologi, Health, Aarhus Universitet
Artikkelen ble først publisert i iTandlægebladet nr. 8, 2016: Hald JD, Farholt S, Gjørup H, Haubek D. Osteogenesis imperfecta - odontologiske implikationer. Tandlægebladet. 2016; 120: 708 - 16.
Korresponderende forfatter: Jannie Dahl Hald, Medicinsk Endokrinologisk Afdeling, Aarhus Universitetshospital, Tage Hansensgade 2, 8000 Aarhus C. Danmark. E-mail: jahald@rm.dk
Accepteret den 20. januar 2016. Artikkelen har gjennomgått ekstern faglig vurdering.
Hald JD, Farholt S, Gjørup H, Haubek D. Osteogenesis imperfecta - odontologiske implikationer. En oversigtsartikel. Nor Tannlegeforen Tid. 2016; 126: 690-7.