
Dysplasi af emalje eller dentin ved medfødt, arvelig sygdom
Klinisk perspektiv
Som kliniker er det vigtigt at være opmærksom på, at visse sjældne sygdomme kan medføre dentale abnormiteter. Disse kan optræde som dysplasi af emalje, dentin eller cement, og de kan medføre behov for ekstraordinære, eventuelt specialiserede, behandlinger hos tandlægen. Såvel sjældne hudsygdomme som sjældne skeletsygdomme kan betinge dentale abnormiteter. Gennem kontakt til de odontologiske videncentre kan tandlægen opnå råd om behandling af disse patienter. Herigennem kan det bl.a. afklares, om det for patienten er relevant at ansøge om særligt tilskud til tandbehandling i privat praksis, eller om patienten bør henvises til et specialiseret behandlingstilbud i videncenterregi.
Dysplasi af dentale væv kan optræde som delsymptom ved sjældne medfødte sygdomme. Oversigtsartiklen beskriver udvalgte eksempler på sjældne, medfødte sygdomme, hvor der optræder dysplasi af dentale væv.
Emaljedysplasi optræder ved hudsygdommene epidermolysis bullosa (EB) og fokal dermal hypoplasi (FDH). EB, junctional type, har hypomineraliseret og hypoplastisk emalje. FDH har hypoplastisk emalje med uregelmæssig overflade og atypisk kronemorfologi. Begge tilstande medfører store pædodontiske og protetiske behandlingsbehov.
Dentindysplasi optræder ved skeletsygdommene osteogenesis imperfecta (OI), der skyldes en kollagen defekt, og X-bunden hypofosfatæmi (XLH), der er en metabolisk knoglesygdom. Dentindysplasi ved OI benævnes dentinogenesis imperfecta (DI) og ses ved svær OI. DI medfører øget risiko for tandfrakturer og tandtab. Ved XLH optræder uregelmæssigheder i hele pulpa-dentin-organet, og der er risiko for spontan udvikling af pulpanekrose. Ved XLH kan også ses elementer af emaljedysplasi (enamel cracks). Cementdysplasi ses ved skeletsygdommen hypofosfatasi (HPP), der optræder i flere alvorlighedsgrader. I varierende grad er tandrodens cementlag acellulær. Der ses præmatur eksfoliering af primære tænder eller ikke-parodontitis-betinget tandtab hos voksne. Svær HPP kan også være associeret med emaljedysplasi.
Konklusion: Emalje- eller dentindysplasi kan optræde ved sjældne sygdomme og medføre ekstraordinære tandbehandlingsbehov. Dette tilsiger henvisning til de odontologiske videncentre med henblik på faglig sparring om diagnostik og behandling.
MED DYSPLASI forstås en abnorm organisering og funktion af cellers udvikling til en given vævstype samt det afvigende morfologiske og strukturelle resultat af disse cellers abnorme funktion [1]. Et eksempel på dysplasi er visse former for tandmisdannelser, hvor et eller flere væv, der specifikt indgår i tanddannelse, fejludvikles. Det kan fx være den genetisk betingede fejlfunktion af ameloblaster eller dentinoblaster, der medfører et tandsæt med henholdsvis amelogenesis imperfecta eller dentinogenesis imperfecta [2]. Disse tilstande behandles i anden artikel i dette nummer af Tandlægebladet. Dysplasi af dentale væv kan imidlertid også forekomme som delsymptom ved sjældne medfødte sygdomme, hvor der er fejludvikling (dysplasi) af flere vævstyper eller organer. Det er formålet med denne artikel at beskrive eksempler på sådanne sjældne, medfødte sygdomme, hvor der optræder dysplasi af dentale væv.
Epidermolysis bullosa
Epidermolysis bullosa (EB) er en gruppe af arvelige sygdomme, hvis fællestræk er skrøbelighed af overfladevæv, først og fremmest hud. Ved alle typer af EB ses tilbagevendende blæredannelser eller erosioner i hud, evt. også slimhinder, men der er stor variation i det kliniske billede af EB. Den histo-patologiske baggrund for hudsymptomerne er brist i den normale binding mellem cellerne i overhuden (epidermis) og den underliggende læderhud (dermis). Der optræder en spaltning (splitting) af basalmembranen i epidermis. Jo dybere beliggende dette »split« er, desto alvorligere symptomer. De sværeste former er svært invaliderende, og kurativ behandling findes ikke [3].
Der beskrives fire overordnede typer af EB, og klassifikationen baseres på lokalisationen af splittet i basalmembranen.
EB simplex
EB simplex karakteriseres ved spaltning øverst i basalmembranen (intraepidermalt) og er oftest med milde symptomer, hvor sårene heler uden ardannelse. Som hovedregel mildnes symptomerne under puberteten. Der er beskrevet et antal forskellige gener, der er associeret med EB simplex, og identificeringen af disse har medført en underopdeling i flere subtyper af EB simplex. EB simplex er normalt ikke associeret med dysplasi af de dentale væv. Til gengæld har relativt mange, specielt ved generaliseret EB simplex, sarte slimhinder med hyppig blisterdannelse i mundhulen. Ophelingen af disse læsioner er som hovedregel uproblematisk, og der optræder ikke væsentlige ardannelser [4].
Junctional EB
Ved junctional EB optræder splittet i overhudens lamina lucida tæt på læderhuden. Som hovedregel er symptomerne vedvarende livet igennem, og hyppigt forekommende infektioner medfører en del ardannelse. Der skelnes mellem lokaliserede former, hvor der alene er vable- og sårdannelser på hænder, fødder, albuer og knæ, og generaliserede former med mere udbredte symptomer, der, udover hårtab, kan inkludere symptomer i spiserør, luftveje og urinveje. Junctional EB er oftest associeret med patologiske varianter af det gen, der koder for proteinet laminin-332 eller det gen, der koder for kollagen XVII. Arvegangen er autosomal recessiv. Ved junctional EB optræder i næsten alle tilfælde bullae og sår på mundhulens slimhinder. Kun sjældent er det ensbetydende med arvævsdannelser [4]. Derudover ses ved denne EB-type også emaljedysplasi i form af generaliseret emaljehypoplasi [5] (Fig. 1). Dette har baggrund i en dysfunktion af emaljens ameloblaster, og klinisk kan denne emaljedysplasi ikke skelnes fra det kliniske billede ved amelogenesis imperfecta (AI). Ligesom ved AI kan der være forskellige varianter af emaljemisdannelsen: nogle med helt tynd og jævn emaljeoverflade og andre med meget uregelmæssig og mere eller mindre pitted overflade, der i svære tilfælde kan maskere de normale konturer af den kliniske krone [6]. Et af de gener, der udtrykkes i ameloblasterne og er associeret med EB junctional (LAMB3), er også associeret med AI hypoplastisk type 1A.
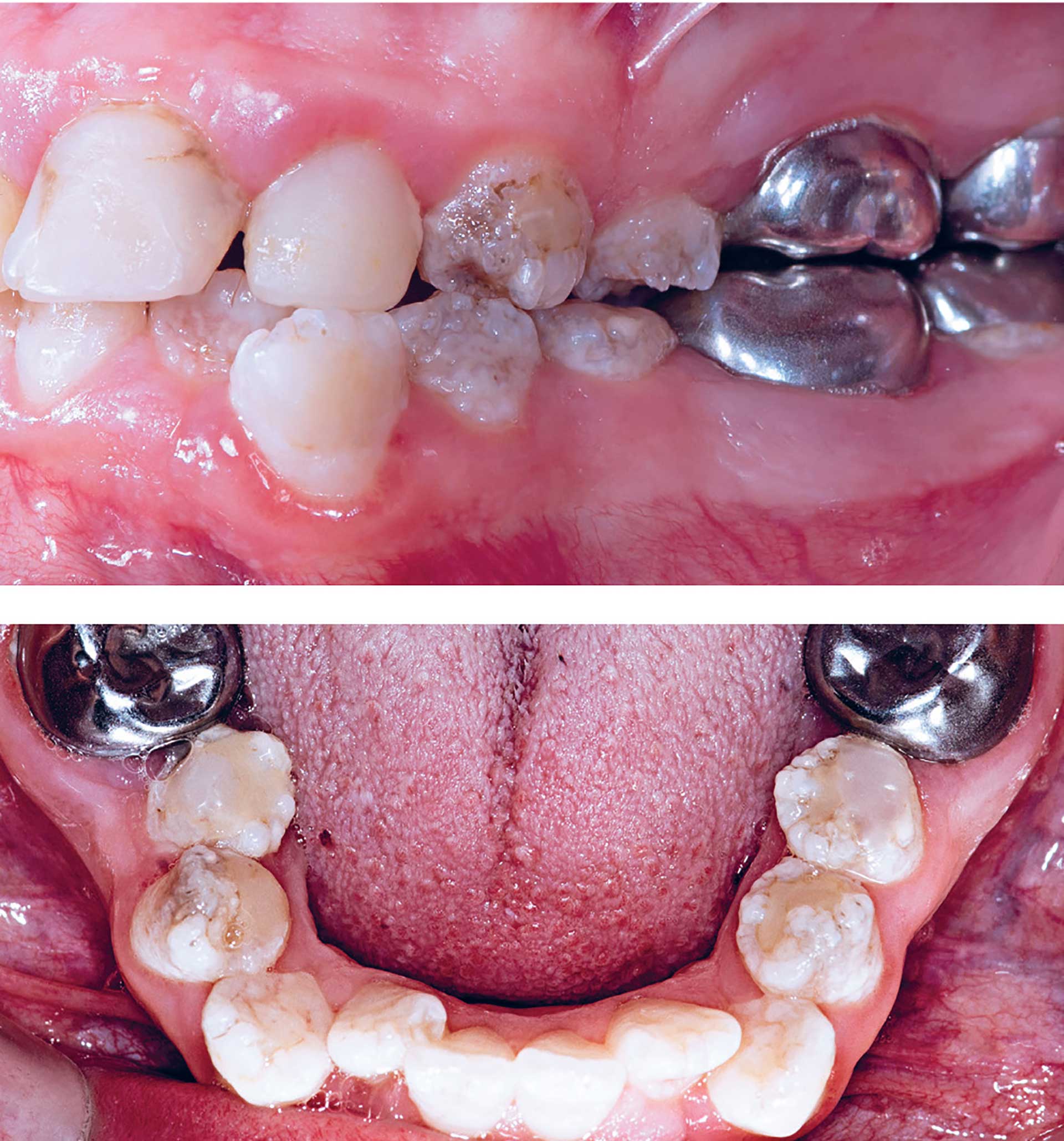
Fig. 1. Pige på 16 år med junctional EB. Tandsæt med svær dysplasi af emaljen med elementer af både hypoplasi og hypomineralisering. Tidligere behandling i form af stålkroner på molarerne, ekstraktion af 14 og 24 orthodontia causa. Incisiver med temporær behandling i form af kompositplast.
Dystrofisk EB
Ved dystrofisk EB optræder splittet endnu dybere, lige under overhudens lamina lucida. Der er adskillige undertyper af den dystrofiske EB, og de er alle associeret med patologiske varianter i genet, der koder for kollagen VII (COL7AI). Arvegangen er autosomal dominant eller recessiv. Nogle af patienterne med den dystrofiske type har en meget vulnerabel hud og kan med tiden udvikle svære ardannelser med sammenvoksning af fingre og tæer samt nedsat bevægelighed af ekstremiteter og eventuelt behov for amputationer. Det er de recessive former, der har de mest alvorlige forløb, og hvor den overordnede prognose er dårligst [7]. Genet COL7AI udtrykkes ikke af ameloblaster, og emaljen kan udvikle sig normalt. Til gengæld kan der i mundhulen optræde svær slimhindeaffektion, som vanskeliggør sufficient renhold af tænder, og hvor der i svære tilfælde udvikles mikrostomi pga. ekstreme arvævsdannelser [6].
Kindlers syndrom
Den 4. hovedtype er Kindlers syndrom synonymt med »mixed EB«, hvor splittet optræder i forskellige niveauer. Arvegangen er autosomal recessiv, og tilstanden er associeret med patologiske varianter af genet KIND1. Der ses ikke afvigelser i emaljelaget. Til gengæld er der beskrevet en association med tidligt debuterende parodontal nedbrydning [6].
Fokal dermal hypoplasi
Fokal dermal hypoplasi (FDH) er en sjælden, syndromal tilstand med afvigelser i såvel ektodermalt som mesodermalt deriverede væv. FDH er også kendt under navnet Goltz syndrom, og som betegnelsen FDH antyder, er der væsentlige hudsymptomer. De klassiske symptomer inkluderer desuden ekstremitetsdefekter, øjenmalformation og dysmorfe ansigtstræk. Abnormiteternei huden optræder som pletvise hudhypoplasier, herniering af subkutant fedt, hypoplastiske negle, sparsom hårvækst samt udslæt i huden omkring mund, næse og øjne. Desuden kan der optræde papillomer i slimhinder [8]. FDH er associeret med patogene varianter (mutationer) af genet PORCN, der sidder på X-kromosomet (Xp11.23), og som har betydning for signalering mellem celler (WNT signaling pathways). Et antal forskellige mutationer og deletioner er beskrevet [9]. Ved FDH er der tale om X-bundet dominant arvegang, hvorfor FDH oftest er letal for drengefostre. Hvis baggrunden for FDH er en postzygotisk mosaicisme (dvs. at kun nogle af fostrets/barnets celler har den patogene variant af PORCN), er også drengebørn levedygtige [10].
FDH vil ofte indebære både kraniofaciale afvigelser og dentale anomalier foruden mere eller mindre iøjnefaldende hudforandringer i ansigtet [11]. De dysmorfe ansigtstræk inkluderer en universel ansigtsasymmetri, en hypoplastisk og uregelmæssig næsefløj, der oftest optræder unilateralt, og morfologiske afvigelser i ydre øres foldninger i forening med, at ørerne er lavt satte og anteverterede. Desuden ses et underudviklet mellemansigt og en markeret hage. Dentitionen er præget af varierende grader af emaljehypoplasi, hvilket medfører dels en uregelmæssig overflade på tandkronerne, dels en afvigende kronemorfologi (Fig. 2). Den afvigende kronemorfologi kan optræde som generaliseret eller regional mikrodonti eller atypisk kronefacon med fx talon cusps, markante hak i incisialkanten (incisal notching) af incisiver og hjørnetænder eller meget irregulære ydre konturer af præmolarer og molarer [12]. Det skal bemærkes, at tændernes emalje på trods af uregelmæssighederne må antages at have normal hårdhed og styrke. Hypodonti er også almindeligt forekommende, og der kan være forsinket eruption af tænder. De dentale symptomer er oftest mere dominerende og alvorlige i den ene side end i den anden side af mundhulen, hvilket reflekterer den asymmetri, der generelt er i symptombilledet ved FDH [12].
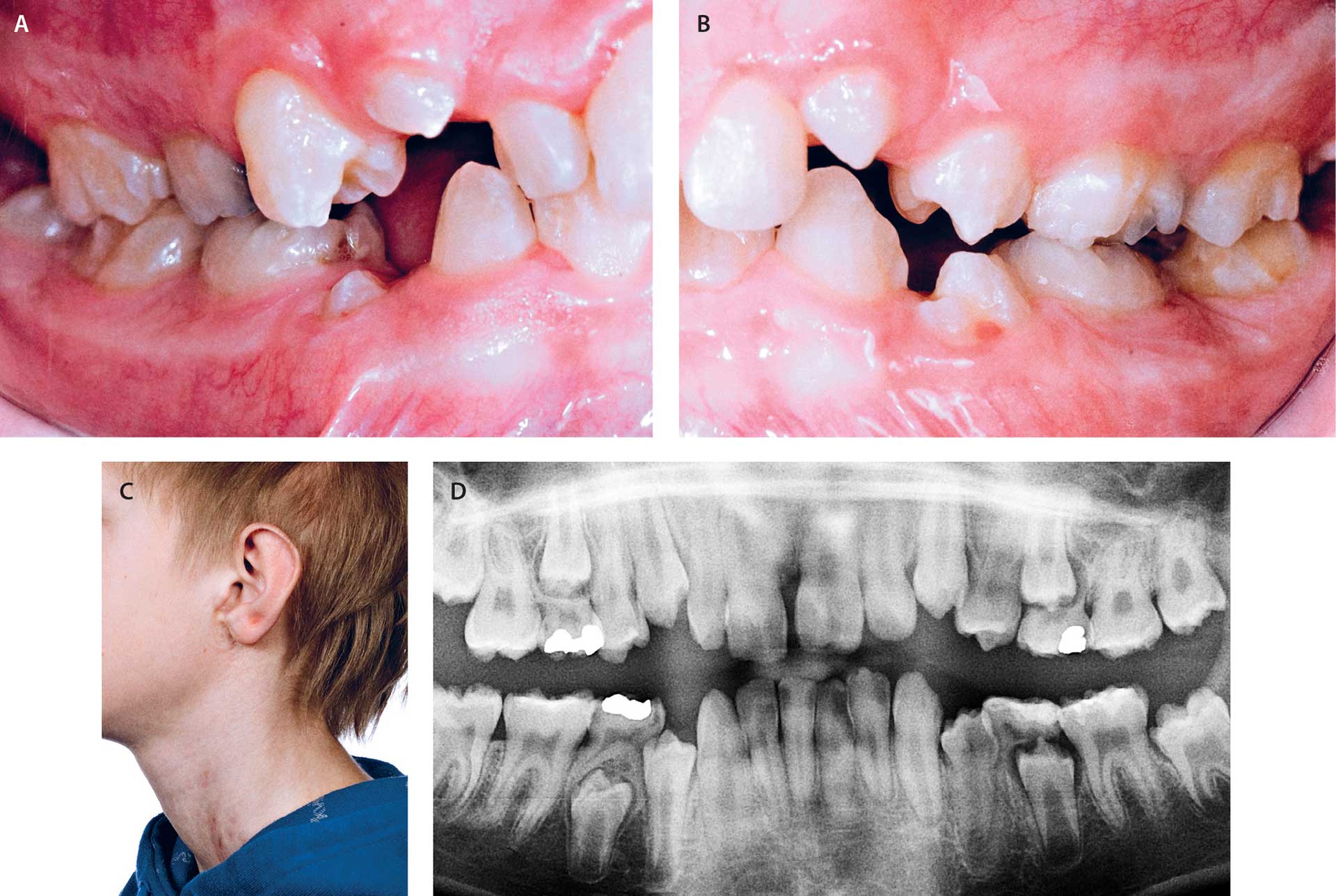
Fig. 2. Dreng på 12 år med FDH (Goltz syndrom). Der er tale om et tilfælde af post-zygotisk mosaicisme.
A-B. Intraorale fotos, der viser mere udtalt emaljehypoplasi i venstre side end i højre side.
C. Venstre side af ansigt og hals, hvor hudsymptomerne er mere udtalte i venstre side end i højre side.
D. Panoramarøntgen, der viser varierende grader af emaljehypoplasi i tandsættet.
Den eksisterende faglitteratur om FDH er sparsom, ikke mindst når det gælder rapportering af tandlægelig behandling. For børn med FDH gælder som for andre, at det er væsentligt at undgå cariesudvikling gennem opretholdelse af en god mundhygiejne. De restorative behandlingsprincipper, der er relevante for børn med FDH, vil i hovedtrækkene være de principper, der gælder for behandling af børn og unge med AI af hypoplastisk type (se dette blads artikel om AI). Som beskrevet ovenfor er emaljen ved FDH netop hypoplastisk, og prognosen for tænder og tandsæt er normalt god, hvis »almindelig« tandsygdom undgås. Der kan opnås god binding af plastmaterialer til emaljen, når der er behov for med kompositplast at udbygge tænder til normal morfologi. I et vist omfang kan der hos den unge voksne også blive behov for kroneprotetisk behandling. I områder med agenesi eller ekstrem grad af mikrodonti kan desuden implantatbaseret protetik blive relevant. Protetiske behandlinger på unge voksne med FDH kan være ganske omfattende og vil med rimelighed kunne tilbydes som et specialiseret behandlingstilbud i de odontologiske videncentre.
Osteogenesis imperfecta
Osteogenesis imperfecta (OI) er en bindevævslidelse, karakteriseret ved knogleskørhed og en øget risiko for low-impactfrakturer [13]. Symptomerne ved OI kan omfatte varierende grader af skeletdeformitet, blå sclerea, høretab og forstyrrelser i tanddannelse. Skeletsmerter og fysisk funktionsnedsættelse forekommer hyppigt [14] [15] [16]. OI er i de fleste tilfælde forårsaget af genetisk betinget defekt i kollagen type 1. Dette protein udgør en væsentlig del af kroppens bindevæv. I Danmark skønnes prævalensen af OI at være 11 pr. 100.000 [17]. Den klassiske OI-klassifikation omfatter fire kliniske undertyper (I-IV): type I (lette og mindre knogledeformiteter), type II (alvorlige knogledeformiteter og perinatal død), type III (meget kort statur og alvorlige, fremadskridende knogledeformiteter) og type IV (mildt påvirket vækst og moderate knogledeformiteter)[18]. OI type I er den hyppigst forekommende type, og denne type er associeret med kvantitative afvigelser i kollagen 1 i modsætning til de øvrige typer, der er karakteriseret ved kvalitative afvigelser i kollagen 1 [13]. Den klassiske opdeling af OI i fire typer er i nyere tid erstattet af mere differentieret klassifikation baseret på det stadigt større kendskab til den genetiske baggrund for OI [19]. Der findes ikke nogen kurativ behandling, og den enkelte patient har en konstant udfordring i at takle sine handikap og funktionsbegrænsninger [20]. Børn med de sværere former for OI modtager antiresorptiv (bisfosfonat) behandling med henblik på at reducere forekomsten af frakturer. De får i mange tilfælde indsat intramedullære stave, der bidrager til udretning af krumme ekstremiteter og så vidt muligt opretholdelse af en gangfunktion. Lavdosis bisfosfonatbehandling ordineres også til en del af voksengruppen, hvor håndtering af skeletsmerter er i fokus.
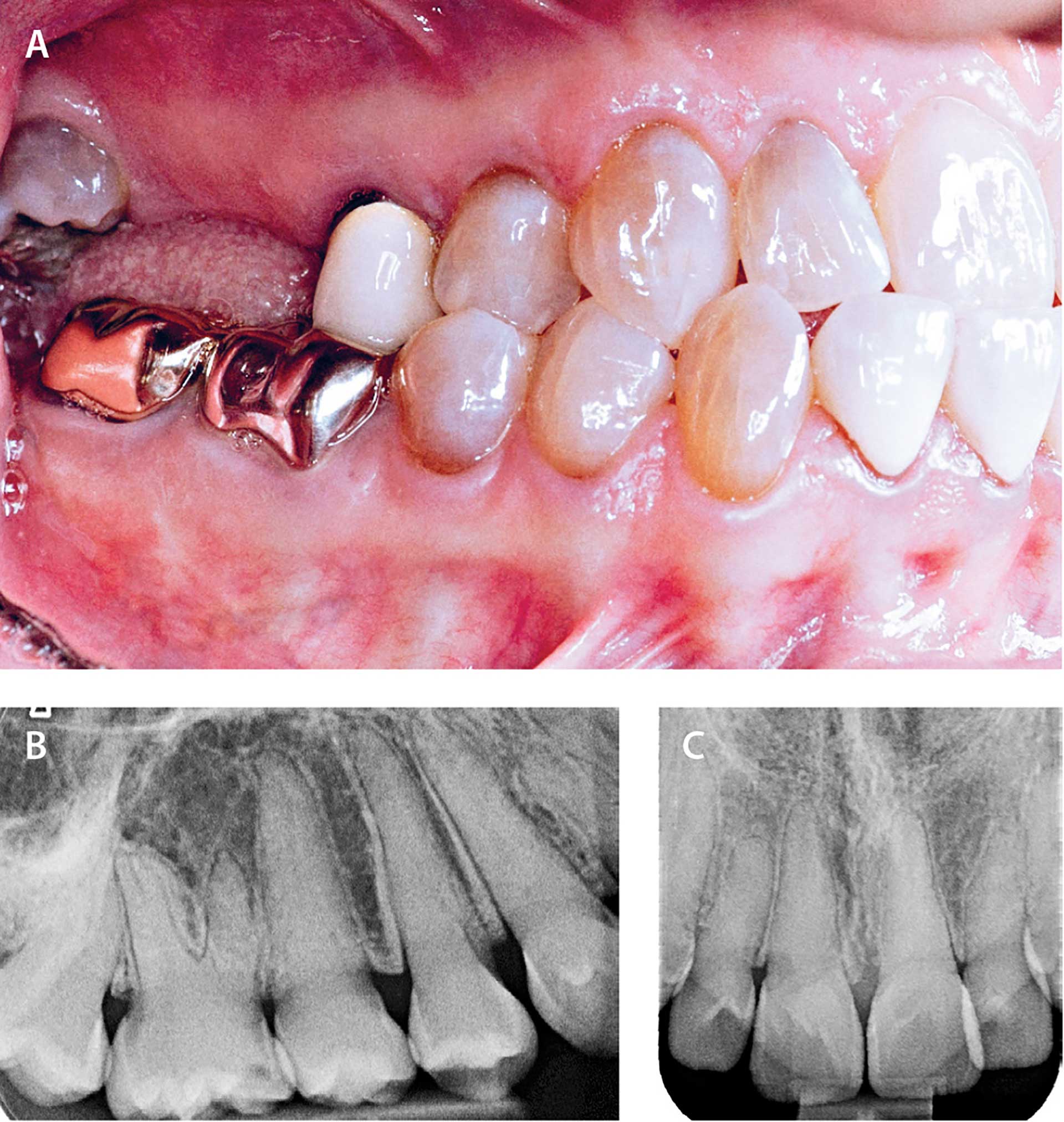
Fig. 3. 45-årig voksen med moderat svær OI og kliniske tegn på dentinogenesis imperfecta (grålig-brunlige farveafvigelser).
A. Tidligere tandtab (17,16) og kronebehandling (15,47,46).
B-C. 20-årig med svær osteogenesis imperfecta og klare radiologiske tegn på dentinogenesis imperfecta (obliteration af pulpacavae
Hos ca. 25 % af personer med OI er der dentale symptomer i form af dentinogenesis imperfecta (DI) [21]. Det er i næsten alle tilfælde personer med de sværere OI-former (OI type III-IV), der også har DI. Ved DI er dentinen dysplastisk pga. de kollagenabnormiteter, der kendetegner OI, enten som et reduceret kollagenindhold (kvantitativ kollagendefekt) eller som fejlkonstrueret kollagen (kvalitativ kollagendefekt), hvor irregulariteten i det histologiske billede af dentinen tiltager med tiltagende sværhedsgrad af OI [22] [23] [24]. Klinisk vil der ved DI ses cervikal konstriktion og pulpaobliteration foruden en gullig, grålig eller brunlig kronefarve (Fig. 3). Tænder med DI anses for i sammenligning med andre tænder at have en øget risiko for kronefrakturer. Dette kan være i form af cervikale kronefrakturer og efterfølgende tandtab [21]. En hyppig dental problemstilling ved de sværere OI-typer er desuden eruptionsvanskeligheder med specielt impakterede anden-molarer [23]. Hos personer med de sværere former for OI ses også en øget forekomst af malokklusion i form af mandibulært overbid og åbent bid [25] [26]. Ortognatkirurgisk behandling kan være aktuel, evt. som osseodistraktion på maksillen, der hos visse kan kræve ganske store avanceringer [27]. Behandling med implantatbaseret protetik ser ud til at kunne gennemføres med en god prognose, dog kun vurderet på et begrænset antal rapporter [28]. Mange børn og voksne med OI bliver nu om dage behandlet med lavdosis antiresorptive medicinske præparater enten som frakturprofylakse eller som led i en reduktion af skeletsmerter. Den bekymring, der kunne være i forhold til evt. induktion af medicinsk betinget osteonekrose i kæberne hos personer med OI, har til nu vist sig delvis ubegrundet. Dog anbefales, at fx tandekstraktioner ikke foretages umiddelbart før eller efter infusion af bisfosfonat [29] [30].
X-bunden hypofosfatæmi
X-bunden hypofosfatæmi (XLH) er karakteriseret ved en utilstrækkelig mineralisering af knogler og tandvæv pga. unormalt tab af fosfat via nyrerne [31]. XLH er den mest almindelige af de arvelige former for hypofosfatæmi, og sygdommen er forårsaget af mutationer i genet, der koder for en fosfatregulerende endopeptidase, der særligt udtrykkes i knogle og dentale væv (PHEX). Hos børn kaldes sygdommen X-bundet hypofosfatæmisk rakitis (XLHR) [32]. I Danmark er prævalensen af XLH 4,8:100.000 [33]. De typiske kliniske træk hos personer med XLH er begrænset legemshøjde, korte underekstremiteter, knoglesmerter og skeletdeformiteter. Bl.a. ses deformering af vægtbærende ekstremiteter (hjulben) og rakitisforandringer i form af hævede metafyser. Voksne med XLH udvikler med alderen ofte mineraliserende entesopatier og slidgigt, begge dele forbundet med smerter. Den traditionelle medicinske behandling består af fosfattilskud og aktivt vitamin D (calcitriol eller alfacalcidol). Siden 2018 har det været muligt at behandle børn med XLH med Burosumab©. Burosumab© er et humant, monoklonalt antistof, som er i stand til at binde Fibroblast Growth Factor 23 (FGF23) og dermed hæmme dets aktivitet. Nedsat FGF23 aktivitet influerer positivt på nyrernes reabsorption af fosfat, og den ugunstige fosfatudskillelse bremses [34]. For nuværende er behandlingen tilgængelig i Danmark for børn og unge fra etårsalderen.
Patienterne med XLH bliver ofte ramt af abscesdannelse på tænder, der tilsyneladende er intakte og uden umiddelbare tegn på patologi [35] [36] [37] [38]. Antallet af endodontisk påvirkede tænder stiger med stigende alder, og hos ældre individer med XLH er det et typisk træk, at alle eller næsten alle tænder i underkæbefronten er rodbehandlede [39] (Fig. 4). Baggrunden for denne udvikling er antagelig en dysplastisk dentin, hvor histologiske studier har påvist et forstørret prædentinlag samt en irregulær og dårligt organiseret dentin med store interglobulære hulrum [40]. Det er dog sandsynligt, at også emaljemisdannelser kan spille en rolle for udvikling af pulpanekrose og infektion ved XLH. Analyser af emaljeoverfladen ved hjælp af scanning elektronmikroskopi (SEM) har demonstreret en irregulær let hypoplastisk emalje hos personer med XLH. Irregulariteten kan have karakter af nogle dybe mikrokløfter ind i emaljen og antagelig være indgangsporten for infektion af de underliggende væv [37].
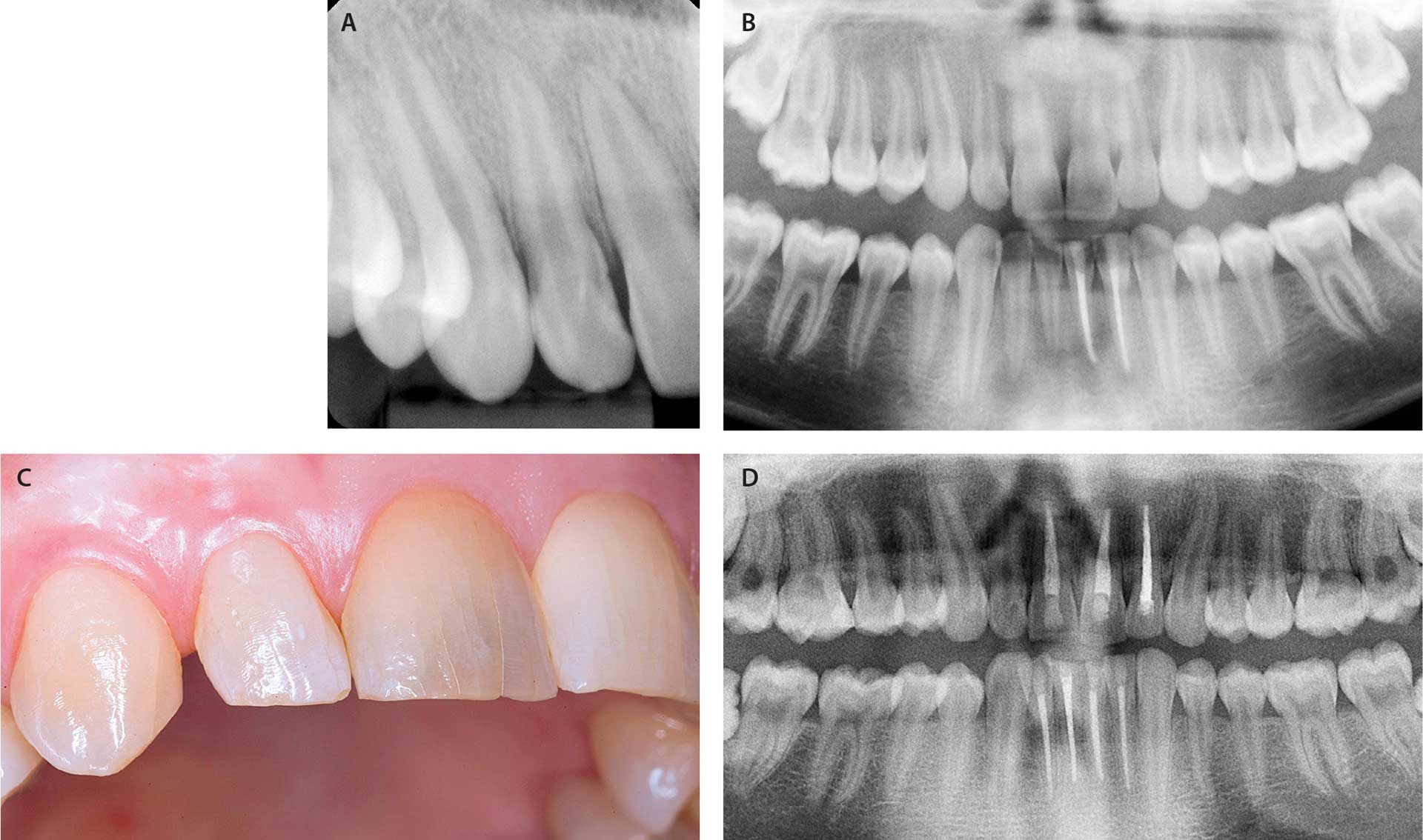
Fig. 4. A-B. Dental røntgen på 13-årig dreng. Store pulpacavae og endodontisk behandlede UK-incisiver (31,32).
C. Samme person som A-B nu 20 år gammel. Bemærk, at der nu er 6 endodontisk behandlede incisiver, og at tænderne derudover er næsten ubehandlede.
D. Incisiver og hjørnetand på en 43-årig kvinde med XLH. Grålig farveafvigelse på 11 og emalje med infraktioner/cracks.
Patienterne med XLH vil ofte have behov for endodontisk behandling [39]. Disse kan gennemføres efter gængse principper, og det mest særegne er det forhold, at den endodontiske behandling ofte skal udføres på tilsyneladende intakte tænder. Da mikroporøsiteter i emaljen antagelig kan disponere for inficering af de underliggende væv, kunne der være en vis logik i at forsegle de kliniske kroners overflade med resin (ekstenderet fissurforsegling). Metoden er tidligere anbefalet af andre [37]. Øjensynligt har behandlingen med Burosumab ikke nogen effekt på de dentale symptomer ved XLH [41].
Hypofosfatasi
Hypofosfatasi (HPP) er en sjælden knoglemetabolisk sygdom, der er karakteriseret ved nedsat aktivitet af det vævsuspecifikke enzym, alkalisk fosfatase (Tissue Non-Specific Alkaline Phosphatase,TNSALP). TNSALP spalter difosfater, hvorefter monofosfater sammen med calcium danner den hydroxylapatit, der er afgørende for mineralisering af knogler og dentale væv. Den nedsatte aktivitet af TNSALP ved HPP bevirker derfor en mangelfuld mineralisering af knogle og tænder. De kliniske symptomer varierer fra meget milde symptomer til ekstremt svære symptomer, der er uforenelige med liv [42]. Traditionelt opereres der med seks forskellige kliniske former for HPP: perinatal HPP, der er den sværeste form og forbundet med høj dødelighed, idet skelettet er uhyre mangelfuldt mineraliseret; infantil HPP, der debuterer før seksmåneders-alderen og medfører dårlig trivsel, muskelsvaghed, kramper, kraniesynostoser, nefrocalcinose samt rakitisforandringer i bl.a. rørknoglernes metafyser; juvenil HPP, der debuterer i perioden fra seksmåneders- til 18-årsalderen, og hvor symptombilledet varierer, men ligner det, der ses ved infantil HPP. Yderligere ses tidligt tab af primære tænder; adult HPP, der debuterer senere, evt. først omkring 50-årsalderen og da optræder med symptomer i form af muskelsvaghed, skeletsmerter, nedsat fysisk formåen, frakturer m.m. [43].
Prævalensen for perinatal og infantil HPP er meget lav (1:300.000), mens mild HPP forekommer relativt hyppigt (1:6.500) [44]. Arvegangen ved de sjældne, perinatale og infantile typer er autosomal recessiv, mens de øvrige, mildere typer af HPP i højere grad har en autosomal dominant arvegang [45]. Den lægelige behandling af patienter med HPP er normalt ikke kurativ, men må tilpasses symptombilledet hos den enkelte patient. I de senere år er der introduceret en mulighed for til yngre personer med HPP at indgive et enzym-substitutionspræparat, Asfotase, som har vist sig at øge knoglemineraliseringen markant og at forbedre overlevelsesmulighederne for de sværest ramte børn med HPP [46].
Det klassiske dentale symptom ved HPP er spontan eksfoliation af primære tænder, typisk startende med underkæbeincisiver. Tænderne kan eksfolieres meget tidligt, 1-3-årsalderen, det sker uden gingival inflammation eller infektion, og der optræder ikke den normale fysiologiske resorption af roden. Fænomenet optræder ved alle HPP-typer og kan ved de mildere former være det første kliniske tegn på HPP [47]. Baggrunden for tandtabet er dysplasi af rodens cementlag, der i varierende grad kan være acellulær [48]. Hos visse patienter med de alvorligste typer af HPP ses også emaljedysplasi i form af svær emaljehypoplasi, ligesom der nogle steder antydes forstyrrelser i dentindannelsen [47] [48] (Fig. 5, A-C). På dental røntgen kan bemærkes et øget volumen af både krone-pulpakammeret og rodkanalerne, og rødderne kan være kortere og mere spinkle end normalt (Fig. 5, D,E). Teenagere og voksne med HPP kan opleve, at også permanente tænder rammes af fæstetab, løsning og i sidste ende tandtab, som ikke er betinget af parodontitis. Det marginale knogleniveau kan selv hos yngre individer med HPP forsænkes ganske dramatisk. En del voksne med HPP vil på denne baggrund kunne få behov for tandprotetisk behandling. Implantatprotetik vil være et attraktivt valg for de fleste. Der eksisterer i PubMed kun rapportering af tre patienttilfælde, og de udviser alle meget tilfredsstillende resultater efter syv års observationstid eller mere [49] [50].
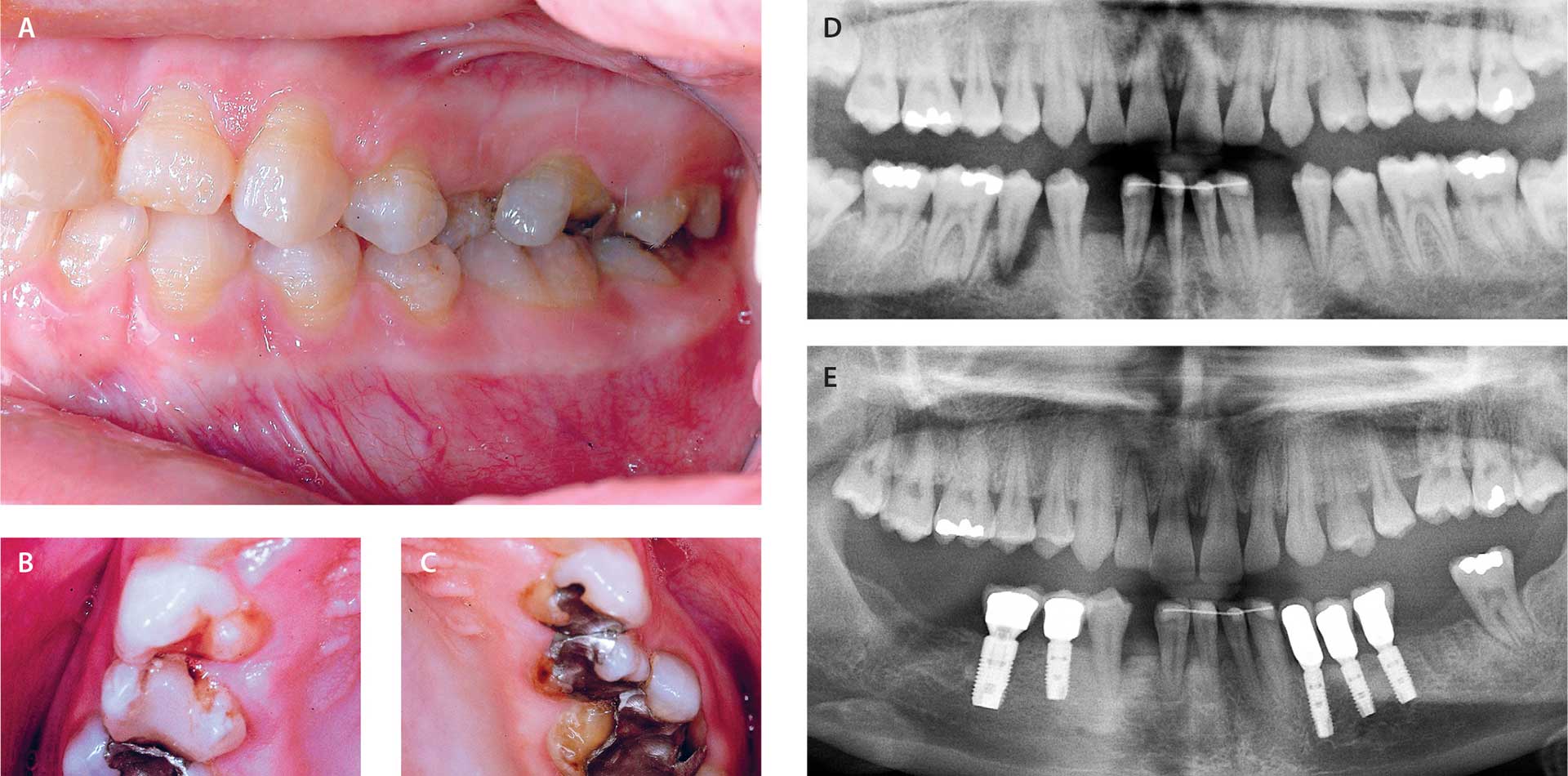
Fig. 5. A-C. 16-årig pige med HPP, hvor der optræder emaljehypoplasi. Adskillige tænder med delvis nedbrydning af tandkrone.
D. 19-årig kvinde med HPP, hvor panoramarøntgen viser agenesi af 33 og 43 samt et generaliseret marginalt fæstetab og total parodontitis på 45.
E. Tandstatus hos samme kvinde som 31-årig. Omfattende tandtab i underkæben, hvor der er indsat implantat-protetiske tanderstatninger.
Konklusion
Emalje- eller dentindysplasi kan optræde ved sjældne sygdomme og kan medføre ekstraordinære tandbehandlingsbehov. Dette tilsiger henvisning til de odontologiske videncentre med henblik på faglig sparring om diagnostik og behandling. Nogle af patienterne med disse tilstande vil have behov for et specialiseret behandlingstilbud, der gennemføres i regi af de odontologiske videncentre. Andre vil kunne få indfriet deres behandlingsbehov i primærsektoren. Denne sidstnævnte gruppe vil i kraft af deres grundlidelse kunne visiteres til et regionalt tandplejetilbud efter sundhedsloven § 166, stk. 3, hvis behandlingsomfanget er ekstraordinært stort og direkte relateret til den sjældne sygdom.
Litteratur
Spranger J, Benirschke K, Hall JG et al. Errors of morphogenesis: concepts and terms. Recommendations of an international working group. J Pediatr. 1982;100:160-5.
Seow WK. Developmental defects of enamel and dentine: challenges for basic science research and clinical management. Austr Dent J. 2014;59 (Supp 1):143-54.
Marchili MR, Spina G, Roversi M et al. Epidermolysis Bullosa in children: the central role of the pediatrician. Orphanet J Rare Dis. 2022;17:147.
Wright JT, Fine JD, Johnson LB. Oral soft tissues in hereditary epidermolysis bullosa. Oral Surg Oral Med Oral Pathol. 1991;71:440-6.
Wright JT, Fine JD, Johnson L. Hereditary epidermolysis bullosa: oral manifestations and dental management. Pediatr Dent. 1993;15:242-8.
Wright JT. Oral manifestations in the epidermolysis bullosa spectrum. Dermatol Clin. 2010;28:159-64.
Fine JD. Inherited epidermolysis bullosa. Orphanet J Rare Dis. 2010;5:12.
Lombardi MP. Focal dermal hypoplasia. Orphanet Enclyclopedia. 2019 [updated April 2019]. (Set 2023 juli). Tilgængelig fra: URL: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=2004&Disease_Disease_Search_diseaseGroup=Focaldermal-hypoplasia&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Focal-dermalhypoplasia&title=Focal%20dermal%20hypoplasia&search=Disease_Search_Simple.
Wang L, Jin X, Zhao X et al. Focal dermal hypoplasia: updates. Oral Dis. 2014;20:17-24.
Happle R. Goltz syndrome and PORCN: A view from Europe. Am J Med Genet C Semin Med Genet 2016;172c:21-3.
Bostwick B, Van den Veyver IB, Sutton VR. Focal dermal hypoplasia. In: Adam MP, Ardinger HH, Pagon RA et al., eds. Seattle (WA): Gene Reviews(®), 1993. University of Washington, Seattle.
Murakami C, de Oliveira Lira Ortega A, Guimarães AS et al. Focal dermal hypoplasia: a case report and literature review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;112:e11-8.
Glorieux FH. Osteogenesis imperfecta. Best Pract Res Clin Rheumatol. 2008;22:85-100.
Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet. 2004;363:1377-85.
Wekre LL, Froslie KF, Haugen L et al. A population-based study of demographical variables and ability to perform activities of daily living in adults with osteogenesis imperfecta. Disabil Rehabil. 2010;32:579-87.
Bishop NJ, Walsh JS. Osteogenesis imperfecta in adults. J Clin Invest. 2014;124:476-7.
Andersen PE, Jr., Hauge M. Osteogenesis imperfecta: a genetic, radiological, and epidemiological study. Clin Genet. 1989;36:250-5.
Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16:101-16.
Mortier GR, Cohn DH, Cormier-Daire V et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019;179:2393-419.
Hald JD, Folkestad L, Harslof T et al. Health-related quality of life in adults with osteogenesis imperfecta. Calcif Tissue Int. 2017;101: 473-8.
Thuesen KJ, Gjørup H, Hald JD et al. The dental perspective on osteogenesis imperfecta in a Danish adult population. BMC Oral Health. 2018;18:175.
Lund AM, Jensen BL, Nielsen LA et al. Dental manifestations of osteogenesis imperfecta and abnormalities of collagen I metabolism. J Craniofac Genet Dev Biol. 1998;18:30-7.
Andersson K, Dahllöf G, Lindahl K et al. Mutations in COL1A1 and COL1A2 and dental aberrations in children and adolescents with osteogenesis imperfecta – A retrospective cohort study. PloS One. 2017;12:e0176466.
Malmgren B, Norgren S. Dental aberrations in children and adolescents with osteogenesis imperfecta. Acta Odontol Scand. 2002;60:65-71.
Bendixen KH, Gjørup H, Baad-Hansen L et al. Temporomandibular disorders and psychosocial status in osteogenesis imperfecta – a cross-sectional study. BMC Oral Health. 2018;18:35.
Waltimo-Sirén J, Kolkka M, Pynnonen S et al. Craniofacial features in osteogenesis imperfecta: a cephalometric study. Am J Med Genet A. 2005;133A:142-50.
Binger T, Rücker M, Spitzer WJ. Dentofacial rehabilitation by osteodistraction, augmentation and implantation despite osteogenesis imperfecta. Int J Oral Maxillofac Surg. 2006;35:559-62.
Oelerich O, Kleinheinz J, Bohner L et al. Dental implants in people with osteogenesis imperfecta: A systematic review. Int J Environ Res Public Health. 2022;19:1563.
Rousseau M, Retrouvey JM. Osteogenesis imperfecta: potential therapeutic approaches. Peer J. 2018;6:e5464.
Hennedige AA, Jayasinghe J, Khajeh J et al. Systematic review on the incidence of bisphosphonate related osteonecrosis of the jaw in children diagnosed with osteogenesis imperfecta. J Oral Maxillofac Res 2014;4:e1.
Beck-Nielsen SS, Brusgaard K, Rasmussen LM et al. Phenotype presentation of hypophosphatemic rickets in adults. Calcif Tissue Int 2010;87:108-19.
THE HYP CONSORTIUM. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet 1995;11:130-6.
Beck-Nielsen SS, Brock-Jacobsen B, Gram J et al. Incidence and prevalence of nutritional and hereditary rickets in southern Denmark. Eur J Endocrinol. 2009;160:491-7.
Imel EA. Burosumab for pediatric X-linked hypophosphatemia. Curr Osteoporos Rep. 2021;19:271-7.
Souza MA, Soares Junior LA, Santos MA et al. Dental abnormalities and oral health in patients with hypophosphatemic rickets. Clinics (Sao Paulo) 2010;65:1023-6.
Baroncelli GI, Angiolini M, Ninni E et al. Prevalence and pathogenesis of dental and periodontal lesions in children with X-linked hypophosphatemic rickets. Eur J Paediatr Dent. 2006;7:61-6.
Cremonesi I, Nucci C, D’Alessandro G et al. X-linked hypophosphatemic rickets: enamel abnormalities and oral clinical findings. Scanning 2014;36:456-61.
Chaussain-Miller C, Sinding C, Wolikow M et al. Dental abnormalities in patients with familial hypophosphatemic vitamin D-resistant rickets: prevention by early treatment with 1-hydroxyvitamin D. J Pediatr. 2003;142:324-31.
Andersen MG, Beck-Nielsen SS, Haubek D et al. Periapical and endodontic status of permanent teeth in patients with hypophosphatemic rickets. J Oral Rehabil. 2012;39:144-50.
Salmon B, Bardet C, Coyac BR et al. Abnormal osteopontin and matrix extracellular phosphoglycoprotein localization, and odontoblast differentiation, in Xlinked hypophosphatemic teeth. Connect Tissue Res. 2014;55 (Supp1):79-82.
Brener R, Zeitlin L, Lebenthal Y et al. Dental health of pediatric patients with X-linked hypophosphatemia (XLH) after three years of burosumab therapy. Front Endocrinol (Lausanne) 2022;13:947814.
Whyte MP. Hypophosphatasia: An overview for 2017. Bone. 2017;102:15-25.
Hepp N, Frederiksen AL, Khosravi J et al. [Diagnostics and treatment of hypophosphatasia]. Ugeskr Laeger. 2018;180: V10170736.
Mornet E, Yvard A, Taillandier A et al. A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann Hum Genet. 2011;75:439-45.
Mornet E. Genetics of hypophosphatasia. Arch Pediatr. 2017;24:5s51-6.
Whyte MP. Hypophosphatasia: Enzyme replacement therapy brings new opportunities and new challenges. J Bone Miner Res. 2017;32:667-75.
Reibel A, Maniere MC, Clauss Fet al. Orodental phenotype and genotype findings in all subtypes of hypophosphatasia. Orphanet J Rare Dis. 2009;4:6.
van den Bos T, Handoko G, Niehof A et al. Cementum and dentin in hypophosphatasia. J Dent Res. 2005;84:1021-5.
Lynch CD, Ziada HM, Buckley LA et al. Prosthodontic rehabilitation of hypophosphatasia using dental implants: a review of the literature and two case reports. J Oral Rehabil. 2009;36:462-8.
Yang Y, Liu Z, Wei L et al. Prosthodontic rehabilitation of a patient with hypophosphatasia using dental implants: A case report with seven years follow-up. J Prosthodont. 2021;30:742-6.
English summary
Dysplasia of dental tissue may appear as a symptom in rare hereditary disease
Dysplasia of dental tissue may appear as a symptom in rare congenital diseases. The present article describes selected examples of rare, congenital diseases in which dysplasia of dental tissues may occur.
Dysplasia of enamel occurs in certain diseases of the skin: epidermolysis bullosa (EB) and focal dermal hypoplasia (FDH). EB, Junctional type, has hypomineralised and hypoplastic enamel. FDH has hypoplastic enamel with an irregular surface and an atypical crown morphology. Both conditions entail major pedodontic and prosthodontic treatment needs. Dysplasia of dentine occurs in certain diseases of the skeleton: osteogenesis imperfecta (OI), which is caused by a collagen defect, and X-linked hypophosphatemia (XLH), which is a metabolic bone disease. In OI, the dental symptom is dentinogenesis imperfecta (DI), which mainly occurs in severe OI. DI leads to an increased risk of tooth fractures and tooth loss. In XLH, irregularities occur in the entire pulp-dentineorgan, and the patients experience a risk of spontaneously evolving necrosis of the pulp. In XLH, elements of enamel dysplasia (enamel cracks) may also be present. Dysplasia of the cementum occurs in another disease of the skeleton: hypophosphatasia (HPP). According to degree of severity, HPP is divided into 6 subtypes. To varying degrees, the cemental layer of the root is acellular. Premature exfoliation of primary teeth may occur. In adults, tooth loss not related to periodontitis may occur. Severe HPP may also be associated with enamel dysplasia.
Conclusion: Enamel or dentine dysplasia can occur in rare diseases and lead to extraordinary dental treatment needs. This requires referral to the dental competence centres for professional advice on diagnostics and treatment.
Korrespondanceansvarlig forfatter: overtandlæge Hans Gjørup, e-post: hans.gjoerup@gmail.com
Accepteret til publikation den 22. juni 2023, og først trykket i Tandlægebladet 2023;127:992-9
Artikkelen er fagfellevurdert
Artikkelen siteres som : Gjørup H. Dysplasi af emalje eller dentin ved medfødt, arvelig sygdom. Nor Tannlegeforen Tid. 2024; 134: 402-10.
Emneord: Rare diseases; Dental enamel; Dentine; Congenital abnormalities
Artikkelen er fagfellevurdert.
Artikkelen siteres som:
Gjørup H. Dysplasi af emalje eller dentin ved medfødt, arvelig sygdom. Nor Tannlegeforen Tid. 2024;134:402-10. doi:10.56373/2024-5-4